Note
Go to the end to download the full example code.
Path integral molecular dynamics¶
- Authors:
Michele Ceriotti @ceriottm
This example shows how to run a path integral molecular dynamics
simulation using i-PI, analyze the output and visualize the
trajectory in chemiscope. It uses LAMMPS
as the driver to simulate the q-TIP4P/f water
model.
import subprocess
import time
import chemiscope
import ipi
import matplotlib.pyplot as plt
import numpy as np
Quantum nuclear effects and path integral methods¶
The Born-Oppenheimer approximation separates the joint quantum mechanical problem for electrons and nuclei into two independent problems. Even though often one makes the additional approximation of treating nuclei as classical particles, this is not necessary, and in some cases (typically when H atoms are present) can add considerable error.
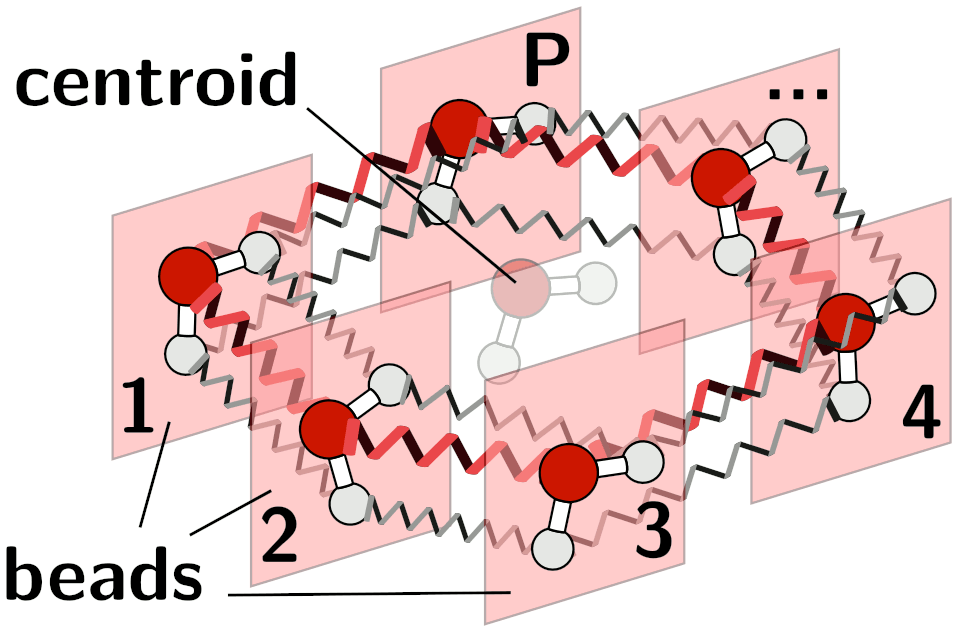
A representation of the ring-polymer Hamiltonian for a water molecule.¶
In order to describe the quantum mechanical nature of light nuclei
(nuclear quantum effects) one of the most widely-applicable methods uses
the path integral formalism to map the quantum partition function of a
set of distinguishable particles onto the classical partition function of
ring polymers composed by multiple beads (replicas) with
corresponding atoms in adjacent replicas being connected by harmonic
springs.
The textbook by Tuckerman
contains a pedagogic introduction to the topic, while
this paper outlines the implementation
used in i-PI.
The classical partition function of the path converges to quantum statistics in the limit of a large number of replicas. In this example, we will use a technique based on generalized Langevin dynamics, known as PIGLET to accelerate the convergence.
Running PIMD calculations with i-PI¶
i-PI is based on a client-server model, with i-PI
controlling the nuclear dynamics (in this case sampling the path Hamiltonian using
molecular dynamics) while the calculation of energies and forces is delegated to
an external client program, in this example LAMMPS.
An i-PI calculation is specified by an XML file.
# Open and read the XML file
with open("data/input_pimd.xml", "r") as file:
xml_content = file.read()
print(xml_content)
<simulation verbosity='medium' safe_stride='100'>
<output prefix='simulation'>
<properties stride='1' filename='out'> [ step, time{picosecond}, conserved{electronvolt}, temperature{kelvin}, kinetic_cv{electronvolt}, potential{electronvolt}, pressure_cv{megapascal}, kinetic_td{electronvolt} ] </properties>
<trajectory filename='pos' stride='20'> positions </trajectory>
<trajectory filename='kin' stride='20'> kinetic_cv </trajectory>
<trajectory filename='kod' stride='20'> kinetic_od </trajectory>
</output>
<total_steps> 200 </total_steps>
<prng>
<seed> 32342 </seed>
</prng>
<ffsocket name='lmpserial' mode='unix' pbc='false'>
<address>h2o-lammps</address> <latency> 1e-4 </latency>
</ffsocket>
<system>
<initialize nbeads='8'>
<file mode='pdb' units='angstrom'> data/water_32.pdb </file>
<velocities mode='thermal' units='kelvin'> 298 </velocities>
</initialize>
<forces>
<force forcefield='lmpserial'> lmpserial </force>
</forces>
<ensemble>
<temperature units='kelvin'>298</temperature>
</ensemble>
<motion mode='dynamics'>
<dynamics mode='nvt'>
<thermostat mode='pile_g'>
<tau units="femtosecond"> 5.0 </tau>
</thermostat>
<timestep units='femtosecond'> 0.5 </timestep>
</dynamics>
</motion>
</system>
</simulation>
NB1: In a realistic simulation you may want to increase the field
total_steps, to simulate at least a few 100s of picoseconds.
NB2: To converge a simulation of water at room temperature, you typically need at least 32 beads. We will see later how to accelerate convergence using a colored-noise thermostat, but you can try to modify the input to check convergence with conventional PIMD
i-PI and lammps should be run separately, and it is possible to launch separate lammps processes to parallelize the evaluation over the beads. On the the command line, this amounts to launching
i-pi data/input_pimd.xml > log &
sleep 2
lmp -in data/in.lmp &
lmp -in data/in.lmp &
Note how i-PI and LAMMPS are completely independent, and
therefore need a separate set of input files. The client-side communication
in LAMMPS is described in the fix_ipi section, that matches the socket
name and mode defined in the ffsocket field in the i-PI file.
We can launch the external processes from a Python script as follows
ipi_process = subprocess.Popen(["i-pi", "data/input_pimd.xml"])
time.sleep(2) # wait for i-PI to start
lmp_process = [subprocess.Popen(["lmp", "-in", "data/in.lmp"]) for i in range(2)]
If you run this in a notebook, you can go ahead and start loading output files before i-PI and lammps have finished running, by skipping this cell
ipi_process.wait()
lmp_process[0].wait()
lmp_process[1].wait()
1
Analyzing the simulation¶
After the simulation has run, you can visualize and post-process the trajectory data. Note that i-PI prints a separate trajectory for each bead, as structural properties can be computed averaging over the configurations of any of the beads.
# drops first frame where all atoms overlap
output_data, output_desc = ipi.read_output("simulation.out")
traj_data = [ipi.read_trajectory(f"simulation.pos_{i}.xyz")[1:] for i in range(8)]
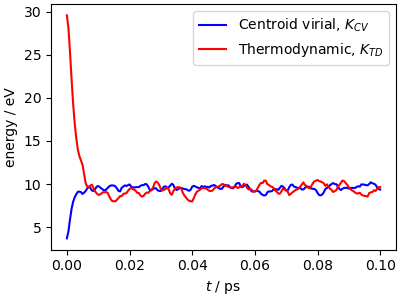
The simulation parameters are pushed at the limits: with the aggressive stochastic thermostatting and the high-frequency normal modes of the ring polymer, there are fairly large fluctuations of the conserved quantity. This is usually not affecting physical observables, but if you see this level of drift in a production run, check carefully for convergence and stability with a reduced time step.
fix, ax = plt.subplots(1, 1, figsize=(4, 3), constrained_layout=True)
ax.plot(
output_data["time"],
output_data["potential"] - output_data["potential"][0],
"b-",
label="Potential, $V$",
)
ax.plot(
output_data["time"],
output_data["conserved"] - output_data["conserved"][0],
"r-",
label="Conserved, $H$",
)
ax.set_xlabel(r"$t$ / ps")
ax.set_ylabel(r"energy / eV")
ax.legend()
plt.show()
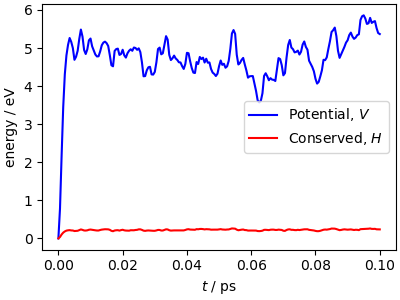
While the potential energy is simply the mean over the beads of the
energy of individual replicas, computing the kinetic energy requires
averaging special quantities that involve also the correlations between beads.
Here we compare two of these estimators: the ‘thermodynamic’ estimator becomes
statistically inefficient when increasing the number of beads, whereas the
‘centroid virial’ estimator remains well-behaved. Note how quickly these estimators
equilibrate to roughly their stationary value, much faster than the equilibration
of the potential energy above. This is thanks to the pile_g thermostat
(see DOI:10.1063/1.3489925) that is
optimally coupled to the normal modes of the ring polymer.
fix, ax = plt.subplots(1, 1, figsize=(4, 3), constrained_layout=True)
ax.plot(
output_data["time"],
output_data["kinetic_cv"],
"b-",
label="Centroid virial, $K_{CV}$",
)
ax.plot(
output_data["time"],
output_data["kinetic_td"],
"r-",
label="Thermodynamic, $K_{TD}$",
)
ax.set_xlabel(r"$t$ / ps")
ax.set_ylabel(r"energy / eV")
ax.legend()
plt.show()
You can also visualize the (very short) trajectory in a way that highlights the fast
spreading out of the beads of the ring polymer. We use chemiscope’s ability to
visualize custom shaped to interleave the trajectories of the beads, forming a
trajectory that shows the connections between the replicas of each atom. Each atom
and its connections are color-coded.
nbeads, nframes, natoms = (
len(traj_data),
len(traj_data[0]),
len(traj_data[0][0]),
)
# creates frames with all beads, so we can use periodic boundary conditions when
# computing distances
full_frames = []
for i in range(nframes):
struc = traj_data[0][i].copy()
for k in range(1, nbeads):
struc += traj_data[k][i]
full_frames.append(struc)
distance_vectors = [
full_frames[frame_i].get_distance(
bead_i * natoms + atom_i,
((bead_i + 1) % nbeads) * natoms + atom_i,
mic=True,
vector=True,
)
for frame_i in range(nframes)
for bead_i in range(nbeads)
for atom_i in range(natoms)
]
paths_shapes = {
"kind": "cylinder",
"parameters": {
"global": {"radius": 0.05},
"atom": [{"vector": d.tolist()} for d in distance_vectors],
},
}
properties = {
"atom_id": [
atom_i
for frame_i in range(nframes)
for bead_i in range(nbeads)
for atom_i in range(natoms)
],
"bead_id": [
bead_i
for frame_i in range(nframes)
for bead_i in range(nbeads)
for atom_i in range(natoms)
],
}
settings = {
"structure": [
{
"atoms": False,
"keepOrientation": True,
"color": {"property": "bead_id", "palette": "hsv (periodic)"},
"bonds": False,
"shape": "paths",
"environments": {"activated": False},
"unitCell": True,
}
]
}
chemiscope.show(
full_frames,
properties=properties,
environments=chemiscope.all_atomic_environments(full_frames, 4.0),
shapes={"paths": paths_shapes},
mode="structure",
settings=settings,
)

Accelerating PIMD with a PIGLET thermostat¶
The simulations in the previous sections are very far from converged – typically one would need approximately 32 replicas to converge a simulation of room-temperature water. To address this problem we will use a method based on generalized Langevin equations, called PIGLET
The input file is input_piglet.xml, that only differs by the definition of
the thermostat, that uses a nm_gle mode in which each normal mode
of the ring polymer is attached to a different colored-noise Generalized Langevin
equation. This makes it possible to converge exactly the simulation results with
a small number of replicas, and to accelerate greatly convergence for realistic
systems such as this. The thermostat parameters can be generated on
the GLE4MD website
ipi_process = subprocess.Popen(["i-pi", "data/input_piglet.xml"])
time.sleep(2) # wait for i-PI to start
lmp_process = [subprocess.Popen(["lmp", "-in", "data/in.lmp"]) for i in range(2)]
ipi_process.wait()
lmp_process[0].wait()
lmp_process[1].wait()
1
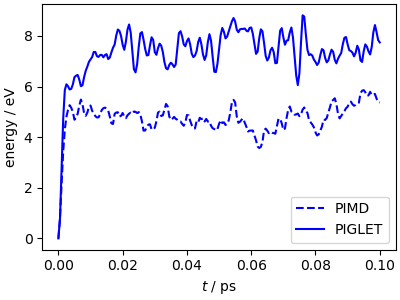
The mean potential energy from the PIGLET trajectory is higher than that for the PIMD one, because it is closer to the converged value (try to run a PIMD trajectory with 64 beads for comparison)
# drops first frame
output_gle, desc_gle = ipi.read_output("simulation_piglet.out")
traj_gle = [ipi.read_trajectory(f"simulation_piglet.pos_{i}.xyz")[1:] for i in range(8)]
fig, ax = plt.subplots(1, 1, figsize=(4, 3), constrained_layout=True)
ax.plot(
output_data["time"],
output_data["potential"] - output_data["potential"][0],
"b--",
label="PIMD",
)
ax.plot(
output_gle["time"],
output_gle["potential"] - output_gle["potential"][0],
"b-",
label="PIGLET",
)
ax.set_xlabel(r"$t$ / ps")
ax.set_ylabel(r"energy / eV")
ax.legend()
plt.show()
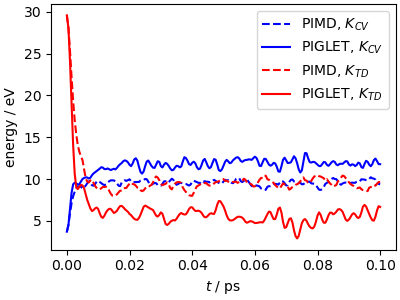
However, you should be somewhat careful: PIGLET converges some but not all the correlations within a path. For instance, it is designed to converge the centroid-virial estimator for the kinetic energy, but not the thermodynamic estimator. For the same reason, don’t try to look at equilibration in terms of the mean temperature: it won’t match the target value, because PIGLET uses a Langevin equation that breaks the classical fluctuation-dissipation theorem, and generates a steady-state distribution that mimics quantum fluctuations.
fix, ax = plt.subplots(1, 1, figsize=(4, 3), constrained_layout=True)
ax.plot(output_data["time"], output_data["kinetic_cv"], "b--", label="PIMD, $K_{CV}$")
ax.plot(output_gle["time"], output_gle["kinetic_cv"], "b", label="PIGLET, $K_{CV}$")
ax.plot(output_data["time"], output_data["kinetic_td"], "r--", label="PIMD, $K_{TD}$")
ax.plot(output_gle["time"], output_gle["kinetic_td"], "r", label="PIGLET, $K_{TD}$")
ax.set_xlabel(r"$t$ / ps")
ax.set_ylabel(r"energy / eV")
ax.legend()
plt.show()
Kinetic energy tensors¶
While we’re at it, let’s do something more complicated (and instructive). Classically, the momentum distribution of any atom is isotropic, so the kinetic energy tensor (KET) \(\mathbf{p}\mathbf{p}^T/2m\) is a constant times the identity matrix. Quantum mechanically, the kinetic energy tensor has more structure, that reflects the higher kinetic energy of particles along directions with stiff bonds. We can compute a moving average of the centroid virial estimator of the KET, and plot it to show the direction of anisotropy. Note that there are some subtleties connected with the evaluation of the moving average, see e.g. DOI:10.1103/PhysRevLett.109.100604
We first need to postprocess the components of the kinetic energy tensors (that i-PI prints out separating the diagonal and off-diagonal bits), averaging them over the last 10 frames and combining them with the centroid configuration from the last frame in the trajectory.
kinetic_cv = ipi.read_trajectory("simulation_piglet.kin.xyz")[1:]
kinetic_od = ipi.read_trajectory("simulation_piglet.kod.xyz")[1:]
kinetic_tens = np.hstack(
[
np.asarray([k.arrays["kinetic_cv"] for k in kinetic_cv[-10:]]).mean(axis=0),
np.asarray([k.arrays["kinetic_od"] for k in kinetic_od[-10:]]).mean(axis=0),
]
)
centroid = traj_gle[-1][-1].copy()
centroid.positions = np.asarray([t[-1].positions for t in traj_gle]).mean(axis=0)
centroid.arrays["kinetic_cv"] = kinetic_tens
We can then view these in chemiscope, setting the proper parameters to
visualize the ellipsoids associated with the KET. Note that some KETs have
negative eigenvalues, because we are averaging over a few frames, which is
insufficient to converge the estimator fully.
ellipsoids = chemiscope.ase_tensors_to_ellipsoids(
[centroid], "kinetic_cv", scale=2, force_positive=True
)
chemiscope.show(
[centroid],
shapes={"kinetic_cv": ellipsoids},
mode="structure",
settings=chemiscope.quick_settings(
structure_settings={
"shape": ["kinetic_cv"],
"unitCell": True,
}
),
)
Total running time of the script: (0 minutes 16.075 seconds)