LAMMPS¶
LAMMPS is a classical molecular dynamics (MD) code, with a focus on materials simulations. It includes a wealth of interatomic potentials, and of basic and advanced molecular simulations techniques, and is highly parallelized using an efficient domain decomposition scheme. Learn more about LAMMPS on its homepage.
Constant-temperature MD and thermostats
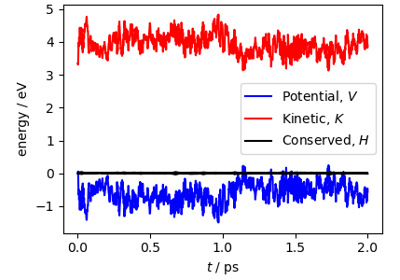
Path integral molecular dynamics
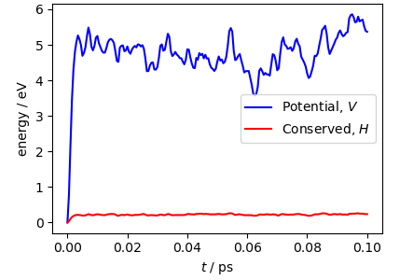
Quantum heat capacity of water
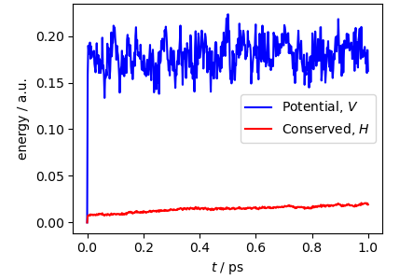
Atomistic Water Model for Molecular Dynamics
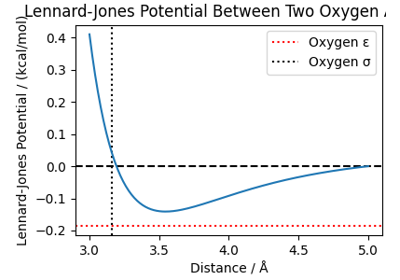
The PET-MAD universal potential
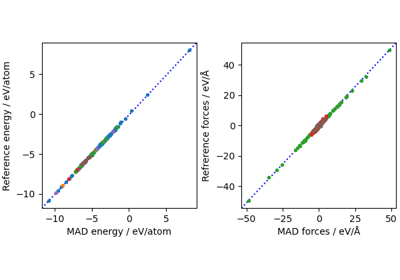
ML collective variables in PLUMED with metatomic
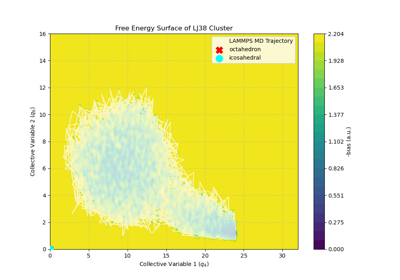
Introduction to foundational models for molecular dynamics
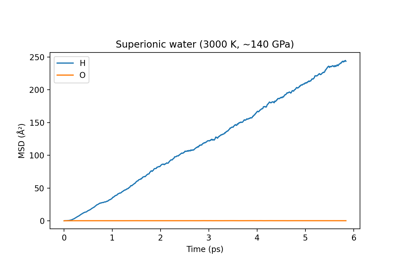
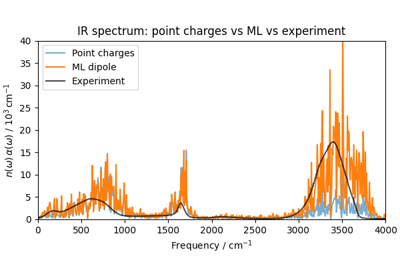
Machine-learned dipoles and infrared spectroscopy of liquid water