Note
Go to the end to download the full example code.
Batch run of CP2K calculations¶
- Authors:
Matthias Kellner @bananenpampe, Philip Loche @PicoCentauri
This is an example how to perform single point calculations based on list of structures
using CP2K using its reftraj functionality. The inputs are a
set of structures in example.xyz using the DFT parameters defined in
reftraj_template.cp2k. The reference DFT parameters are taken from Cheng et
al. Ab initio thermodynamics of liquid and solid water 2019. Due to the small size of the test
structure and convergence issues, we have decreased the size of the CUTOFF_RADIUS
from \(6.0\,\mathrm{Å}\) to \(3.0\,\mathrm{Å}\). For actual production
calculations adapt the template!
We start the example by importing the required packages.
import os
import platform
import subprocess
from typing import List, Union
import ase.io
import ase.visualize.plot
import matplotlib.pyplot as plt
import numpy as np
from atomistic_cookbook_utils import download_with_retry, run_command
Install CP2K¶
We’ll need a working installation of cp2k. The best way to do so depends on your platform, here are some possible solutions, but feel free to replace them with another installation method.
if platform.system() == "Linux":
# use conda on Linux
run_command(
"conda install cp2k=2024.2=openblas_nompi_hbd0aaf2_1000 -c conda-forge -y"
)
elif platform.system() == "Darwin":
# use homebrew on macOS
run_command("brew install cp2k")
else:
print("no known way to install cp2k, skipping installation")
Define necessary functions¶
Next we below define necessary helper functions to run the example.
def write_reftraj(fname: str, frames: Union[ase.Atoms, List[ase.Atoms]]) -> None:
"""Writes a list of ase atoms objects to a reference trajectory.
A reference trajectory is the CP2K compatible format for the computation of batches.
All frames must have the stoichiometry/composition.
"""
if isinstance(frames, ase.Atoms):
frames = [frames]
out = ""
for i, atoms in enumerate(frames):
if (
len(atoms) != len(frames[0])
or atoms.get_chemical_formula() != frames[0].get_chemical_formula()
):
raise ValueError(
f"Atom symbols in frame {i},{atoms.get_chemical_formula()} are "
f"different compared to initial frame "
f"{frames[0].get_chemical_formula()}. "
"CP2K does not support changing atom types within a reftraj run!"
)
out += f"{len(atoms):>8}\n i = {i + 1:>8}, time = {0:>12.3f}\n"
for atom in atoms:
pos = atom.position
out += f"{atom.symbol}{pos[0]:24.15f}{pos[1]:24.15f}{pos[2]:24.15f}\n"
out += "\n"
with open(fname, "w") as f:
f.write(out)
def write_cellfile(fname: str, frames: Union[ase.Atoms, List[ase.Atoms]]) -> None:
"""Writes a cellfile for a list of ``ase.Atoms``.
A Cellfile accompanies a reftraj containing the cell parameters.
"""
if isinstance(frames, ase.Atoms):
frames = [frames]
out = (
"# "
"Step "
"Time [fs] "
"Ax [Angstrom] "
"Ay [Angstrom] "
"Az [Angstrom] "
"Bx [Angstrom] "
"By [Angstrom] "
"Bz [Angstrom] "
"Cx [Angstrom] "
"Cy [Angstrom] "
"Cz [Angstrom] "
"Volume [Angstrom^3]\n"
)
for i, atoms in enumerate(frames):
out += f"{i + 1:>8}{0:>12.3f}"
out += "".join([f"{c:>20.10f}" for c in atoms.cell.flatten()])
out += f"{atoms.cell.volume:>25.10f}"
out += "\n"
with open(fname, "w") as f:
f.write(out)
def write_cp2k_in(
fname: str, project_name: str, last_snapshot: int, cell: List[float]
) -> None:
"""Writes a cp2k input file from a template.
Importantly, it writes the location of the basis set definitions,
determined from the path of the system CP2K install to the input file.
"""
cp2k_in = open("reftraj_template.cp2k", "r").read()
cp2k_in = cp2k_in.replace("//PROJECT//", project_name)
cp2k_in = cp2k_in.replace("//LAST_SNAPSHOT//", str(last_snapshot))
cp2k_in = cp2k_in.replace("//CELL//", " ".join([f"{c:.6f}" for c in cell]))
with open(fname, "w") as f:
f.write(cp2k_in)
We will now download basis set files from CP2K website. Depending on your CP2K installation, this might not be necessary!
for file in ["GTH_BASIS_SETS", "BASIS_ADMM", "POTENTIAL", "dftd3.dat", "t_c_g.dat"]:
download_with_retry(
f"https://raw.githubusercontent.com/cp2k/cp2k/support/v2024.1/data/{file}",
os.path.join("parameters", file),
)
Prepare calculation inputs¶
During this example we will create a directory named project_directory containing
the subdirectories for each stoichiometry. This is necessary, because CP2K can only
run calculations using a fixed stoichiometry at a time, using its reftraj
functionality.
Below we define the general information for the CP2K run. This includes the reference
files for the structures, the project_name used to build the name of the
trajectory during the CP2K run, the project_directory where we store all
simulation output as well as the path write_to_file which is the name of the file
containing the computed energies and forces of the simulation.
frames_full = ase.io.read("example.xyz", ":")
project_name = "test_calcs" # name of the global PROJECT
project_directory = "production"
write_to_file = "out.xyz"
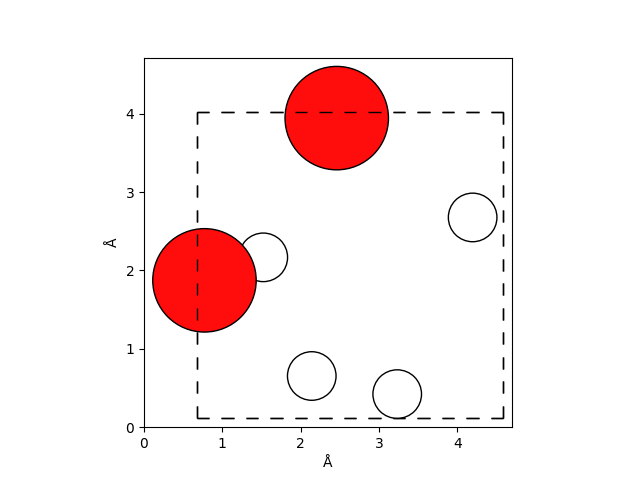
Below we show the initial configuration of two water molecules in a cubic box with a side length of \(\approx 4\,\mathrm{Å}\).
ase.visualize.plot.plot_atoms(frames_full[0])
plt.xlabel("Å")
plt.ylabel("Å")
plt.show()
We now extract the stoichiometry from the input dataset using ASE’s
ase.symbols.Symbols.get_chemical_formula() method.
frames_dict = {}
for atoms in frames_full:
chemical_formula = atoms.get_chemical_formula()
try:
frames_dict[chemical_formula]
except KeyError:
frames_dict[chemical_formula] = []
frames_dict[chemical_formula].append(atoms)
Based on the stoichiometries we create one calculation subdirectories for the
calculations. (reftraj, input and cellfile). For our example this is only is one
directory named H4O2 because our dataset consists only of a single structure with
two water molecules.
for stoichiometry, frames in frames_dict.items():
current_directory = f"{project_directory}/{stoichiometry}"
os.makedirs(current_directory, exist_ok=True)
write_cp2k_in(
f"{current_directory}/in.cp2k",
project_name=project_name,
last_snapshot=len(frames),
cell=frames[0].cell.diagonal(),
)
ase.io.write(f"{current_directory}/init.xyz", frames[0])
write_reftraj(f"{current_directory}/reftraj.xyz", frames)
write_cellfile(f"{current_directory}/reftraj.cell", frames)
Run simulations¶
Now we have all ingredients to run the simulations. Below we call the bash script
run_calcs.sh.
#! /bin/bash
for i in $(find ./production/ -mindepth 1 -type d); do
cd $i
cp2k.ssmp -i in.cp2k
cd -
done
This script will loop through all stoichiometry subdirectories and call the CP2K engine.
# run the bash script directly from this script
subprocess.run("bash run_calcs.sh", shell=True)
CompletedProcess(args='bash run_calcs.sh', returncode=0)
Note
For a usage on an HPC environment you can parallelize the loop over the sub-directories and submit and single job per stoichiometry.
Load results¶
After the simulation we load the results and perform a unit version from the default CP2K output units (Bohr and Hartree) to Å and eV.
cflength = 0.529177210903 # Bohr -> Å
cfenergy = 27.211386245988 # Hartree -> eV
cfforce = cfenergy / cflength # Hartree/Bohr -> eV/Å
Finally, we store the results as ase.Atoms in the new_frames list and
write them to the project_directory using the new_fname. Here it will be
written to production/out_dft.xyz.
new_frames = []
for stoichiometry, frames in frames_dict.items():
current_directory = f"{project_directory}/{stoichiometry}"
frames_dft = ase.io.read(f"{current_directory}/{project_name}-pos-1.xyz", ":")
forces_dft = ase.io.read(f"{current_directory}/{project_name}-frc-1.xyz", ":")
cell_dft = np.atleast_2d(np.loadtxt(f"{current_directory}/{project_name}-1.cell"))[
:, 2:-1
]
for i_atoms, atoms in enumerate(frames_dft):
frames_ref = frames[i_atoms]
# Check consistent positions
if not np.allclose(atoms.positions, frames_ref.positions):
raise ValueError(f"Positions in frame {i_atoms} are not the same.")
# Check consistent cell
if not np.allclose(frames_ref.cell.flatten(), cell_dft[i_atoms]):
raise ValueError(f"Cell dimensions in frame {i_atoms} are not the same.")
atoms.info["E"] *= cfenergy
atoms.pbc = True
atoms.cell = frames_ref.cell
atoms.set_array("forces", cfforce * forces_dft[i_atoms].positions)
new_frames += frames_dft
new_fname = f"{os.path.splitext(os.path.basename(write_to_file))[0]}_dft.xyz"
ase.io.write(f"{project_directory}/{new_fname}", new_frames)
Total running time of the script: (0 minutes 34.844 seconds)