Note
Go to the end to download the full example code.
Atomistic Water Model for Molecular Dynamics¶
- Authors:
Philip Loche @PicoCentauri, Marcel Langer @sirmarcel and Michele Ceriotti @ceriottm
In this example, we demonstrate how to construct a metatomic model for flexible three and four-point water
model, with parameters optimized for use together with quantum-nuclear-effects-aware
path integral simulations (cf. Habershon et al., JCP (2009)). The model also demonstrates the use of
torch-pme, a Torch library for long-range interactions, and uses the resulting model
to perform demonstrative molecular dynamics simulations.
# sphinx_gallery_thumbnail_number = 3
from typing import Dict, List, Optional, Tuple
import ase.io
from atomistic_cookbook_utils import run_command
# Simulation and visualization tools
import chemiscope
import matplotlib.pyplot as plt
# Model wrapping and execution tools
import numpy as np
import torch
# Core atomistic libraries
import torchpme
from ase.optimize import LBFGS
from ipi.scripting import (
InteractiveSimulation,
forcefield_xml,
motion_nvt_xml,
simulation_xml,
read_output,
read_trajectory,
)
from metatensor.torch import Labels, TensorBlock, TensorMap
from metatomic.torch import (
AtomisticModel,
ModelCapabilities,
ModelEvaluationOptions,
ModelMetadata,
ModelOutput,
NeighborListOptions,
System,
load_atomistic_model,
)
# Integration with ASE calculator for metatomic models
from metatomic_ase import MetatomicCalculator
from vesin.metatomic import NeighborList
The q-TIP4P/F Model¶
The q-TIP4P/F model uses simple (quasi)-harmonic terms to describe intra-molecular
flexibility - with the use of a quartic term being a specific feature used to describe
the covalent bond softening for a H-bonded molecule - a Lennard-Jones term describing
dispersion and repulsion between O atoms, and an electrostatic potential between
partial charges on the H atoms and the oxygen electron density. For a four-point
model, the oxygen charge is slightly displaced from the oxygen’s position, improving
properties like the dielectric constant. The
fourth point, referred to as M, is implicitly derived from the other atoms of each
water molecule.
Intra-molecular interactions¶
The molecular bond potential is usually defined as a harmonic potential of the form
Here, \(k_\mathrm{bond}\) is the force constant and \(r_0\) is the equilibrium distance. Bonded terms like this require defining a topology, i.e. a list of pairs of atoms that are actually bonded to each other.
q-TIP4P/F doesn’t use a harmonic potential but a quartic approximation of a Morse potential, that allows describing the anharmonicity of the O-H covalent bond, and how the mean distance changes due to zero-point fluctuations and/or hydrogen bonding.
Note
The harmonic coefficient is related to the coefficients in the original paper by \(k_r=2 D_r \alpha_r^2\).
def bond_energy(
distances: torch.Tensor,
coefficient_k: torch.Tensor,
coefficient_alpha: torch.Tensor,
coefficient_r0: torch.Tensor,
) -> torch.Tensor:
"""Harmonic potential for bond terms."""
dr = distances - coefficient_r0
alpha_dr = dr * coefficient_alpha
return 0.5 * coefficient_k * dr**2 * (1 - alpha_dr + alpha_dr**2 * 7 / 12)
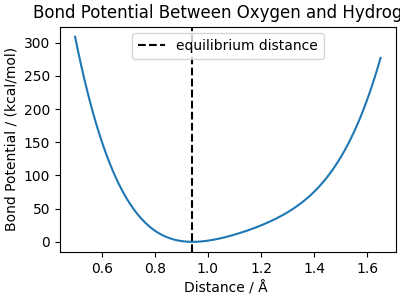
The parameters reproduce the form of a Morse potential close to the minimum, but avoids the dissociation of the bond, due to the truncation to the quartic term. These are the parameters used for q-TIP4P/F
OH_kr = 116.09 * 2 * 2.287**2 # kcal/mol/Å**2
OH_r0 = 0.9419 # Å
OH_alpha = 2.287 # 1/Å
bond_distances = np.linspace(0.5, 1.65, 200)
bond_potential = bond_energy(
distances=bond_distances,
coefficient_k=OH_kr,
coefficient_alpha=OH_alpha,
coefficient_r0=OH_r0,
)
fig, ax = plt.subplots(1, 1, figsize=(4, 3), constrained_layout=True)
ax.set_title("Bond Potential Between Oxygen and Hydrogen")
ax.plot(bond_distances, bond_potential)
ax.axvline(OH_r0, label="equilibrium distance", color="black", linestyle="--")
ax.set_xlabel("Distance / Å ")
ax.set_ylabel("Bond Potential / (kcal/mol)")
ax.legend()
fig.show()
The harmonic angle potential describe the bending of the HOH angle, and is usually modeled as a (curvilinear) harmonic term, defined based on the angle
where \(k_\mathrm{angle}\) is the force constant and \(\theta_0\) is the equilibrium angle between the three atoms.
def bend_energy(
angles: torch.Tensor, coefficient: torch.Tensor, equilibrium_angle: torch.Tensor
):
"""Harmonic potential for angular terms."""
return 0.5 * coefficient * (angles - equilibrium_angle) ** 2
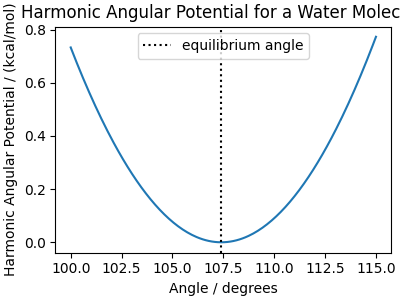
We use the following parameters:
HOH_angle_coefficient = 87.85 # kcal/mol/rad^2
HOH_equilibrium_angle = 107.4 * torch.pi / 180 # radians
We can plot the bend energy as a function of the angle that is, unsurprisingly, parabolic around the equilibrium angle
angle_distances = np.linspace(100, 115, 200)
angle_potential = bend_energy(
angles=angle_distances * torch.pi / 180,
coefficient=HOH_angle_coefficient,
equilibrium_angle=HOH_equilibrium_angle,
)
fig, ax = plt.subplots(1, 1, figsize=(4, 3), constrained_layout=True)
ax.set_title("Harmonic Angular Potential for a Water Molecule")
ax.plot(angle_distances, angle_potential)
ax.axvline(
HOH_equilibrium_angle * 180 / torch.pi,
label="equilibrium angle",
color="black",
linestyle=":",
)
ax.set_xlabel("Angle / degrees")
ax.set_ylabel("Harmonic Angular Potential / (kcal/mol)")
ax.legend()
fig.show()
Lennard-Jones Potential¶
The Lennard-Jones (LJ) potential describes the interaction between a pair of neutral atoms or molecules, balancing dispersion forces at longer ranges and repulsive forces at shorter ranges. The LJ potential is defined as:
where \(\epsilon\) is the depth of the potential well and \(\sigma\) the distance at which the potential is zero. For water there is usually only an oxygen-oxygen Lennard-Jones potential.
We implement the Lennard-Jones potential as a function that takes distances, along
with the parameters sigma, epsilon, and cutoff that indicates the distance
at which the interaction is assumed to be zero. To ensure that there is no
discontinuity an offset is included to shift the curve so it is zero at the cutoff
distance.
def lennard_jones_pair(
distances: torch.Tensor,
sigma: torch.Tensor,
epsilon: torch.Tensor,
cutoff: torch.Tensor,
):
"""Shifted Lennard-Jones potential for pair terms."""
c6 = (sigma / distances) ** 6
c12 = c6**2
lj = 4 * epsilon * (c12 - c6)
sigma_cutoff_6 = (sigma / cutoff) ** 6
offset = 4 * epsilon * sigma_cutoff_6 * (sigma_cutoff_6 - 1)
return lj - offset
We plot this potential to visualize its behavior, using q-TIP4P/F parameters. To highlight the offset, we use a cutoff of 5 Å instead of the usual 7 Å.
O_sigma = 3.1589 # Å
O_epsilon = 0.1852 # kcal/mol
cutoff = 5.0 # Å <- cut short, to highlight offset
lj_distances = np.linspace(3, cutoff, 200)
lj_potential = lennard_jones_pair(
distances=lj_distances, sigma=O_sigma, epsilon=O_epsilon, cutoff=cutoff
)
fig, ax = plt.subplots(1, 1, figsize=(4, 3), constrained_layout=True)
ax.set_title("Lennard-Jones Potential Between Two Oxygen Atoms")
ax.axhline(0, color="black", linestyle="--")
ax.axhline(-O_epsilon, color="red", linestyle=":", label="Oxygen ε")
ax.axvline(O_sigma, color="black", linestyle=":", label="Oxygen σ")
ax.plot(lj_distances, lj_potential)
ax.set_xlabel("Distance / Å")
ax.set_ylabel("Lennard-Jones Potential / (kcal/mol)")
ax.legend()
fig.show()
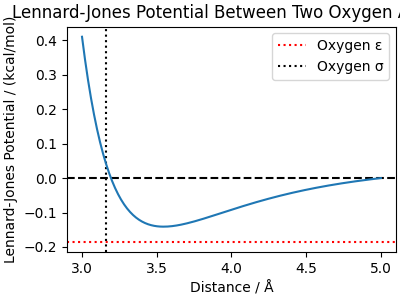
Due to our reduced cutoff the minimum of the blue line does not touch the red horizontal line.
Electrostatic Potential¶
The long-ranged nature of electrostatic interactions makes computing them in simulations non-trivial. For periodic systems the Coulomb energy is given by:
The sum over \(\boldsymbol n\) takes into account the periodic images of the charges and the prime indicates that in the case \(i=j\) the term \(n=0\) must be omitted. Further \(\boldsymbol r_{ij} = \boldsymbol r_i - \boldsymbol r_j\) and \(\boldsymbol L\) is the length of the (cubic)simulation box.
Since this sum is conditionally convergent it isn’t computable using a direct sum. Instead the Ewald summation, published in 1921, remains a foundational method that effectively defines how to compute the energy and forces of such systems. To further speed the methods, mesh based algorithm suing fast Fourier transformation have been developed, such as the Particle-Particle Particle-Mesh (P3M) algorithm. For further details we refer to a paper by Deserno and Holm.
We use a Torch implementation of the P3M method within the torch-pme package.
The core class we use is the torchpme.P3MCalculator class - you can read more
and see specific examples in the torchpme documentation As a demonstration we use two
charges in a cubic cell, computing the electrostatic energy as a function of distance
O_charge = -0.84
coulomb_distances = torch.linspace(0.5, 9.0, 50)
cell = torch.eye(3) * 10.0
We also use the parameter-tuning functionality of torchpme, to provide efficient
evaluation at the desired level of accuracy. This is achieved calling
torchpme.tuning.tune_p3m() on a template of the target structure
charges = torch.tensor([O_charge, -O_charge]).unsqueeze(-1)
positions_coul = torch.tensor([[0.0, 0.0, 0.0], [4.0, 0.0, 0.0]])
neighbor_indices = torch.tensor([[0, 1]])
neighbor_distances = torch.tensor([4.0])
p3m_smearing, p3m_parameters, _ = torchpme.tuning.tune_p3m(
charges,
cell,
positions_coul,
cutoff,
neighbor_indices,
neighbor_distances,
accuracy=1e-4,
)
p3m_prefactor = torchpme.prefactors.kcalmol_A
The hydrogen charge is derived from the oxygen charge as \(q_H = -q_O/2\). The
smearing and mesh_spacing parameters are the central parameters for P3M and
are crucial to ensure the correct energy calculation. We now compute the electrostatic
energy between two point charges using the P3M algorithm.
p3m_calculator = torchpme.P3MCalculator(
potential=torchpme.CoulombPotential(p3m_smearing, prefactor=p3m_prefactor),
**p3m_parameters,
)
For the inference, we need a neighbor list and distances which we compute “manually”. Typically, the neighbors are provided by the simulation engine.
neighbor_indices = torch.tensor([[0, 1]])
potential = torch.zeros_like(coulomb_distances)
for i_dist, dist in enumerate(coulomb_distances):
positions_coul = torch.tensor([[0.0, 0.0, 0.0], [dist, 0.0, 0.0]])
charges = torch.tensor([O_charge, -O_charge]).unsqueeze(-1)
neighbor_distances = torch.tensor([dist])
potential[i_dist] = p3m_calculator.forward(
positions=positions_coul,
cell=cell,
charges=charges,
neighbor_indices=neighbor_indices,
neighbor_distances=neighbor_distances,
)[0]
We plot the electrostatic potential between two point charges.
fig, ax = plt.subplots(1, 1, figsize=(4, 3), constrained_layout=True)
ax.set_title("Electrostatic Potential Between Two Point Charges")
ax.plot(coulomb_distances, potential)
ax.set_xlabel("Distance / Å")
ax.set_ylabel("Electrostatic Potential / (kcal/mol)")
fig.show()
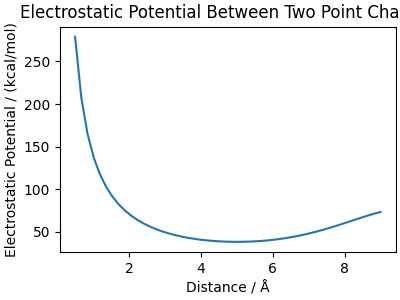
The potential shape may appear unusual due to computations within a periodic box. For small distances, the potential behaves like \(1/r\), but it increases again as charges approach across periodic boundaries.
Note
In most water models, Coulomb interactions within each molecule are excluded, as intramolecular energies are already parametrized by the bond and angle interactions. Therefore, in our model defined below we first compute the electrostatic energy of all atoms and then subtract interactions between bonded atoms.
Implementation as a TIP4P/f torch module¶
In order to implement a Q-TIP4P/f potential in practice, we first build a class that
follows the interface of a metatomic model. This requires defining the atomic
structure in terms of a metatomic.torch.System object - a simple
container for positions, cell, and atomic types, that can also be enriched with one or
more metatomic.torchist objects holding neighbor
distance information. This is usually provided by the code used to perform a
simulation, but can be also computed explicitly using ase or vesin, as we do here.
For running our simulation we use a small waterbox containing 32 water molecules.
atoms = ase.io.read("data/water_32.xyz")
chemiscope.show(
[atoms],
mode="structure",
settings=chemiscope.quick_settings(structure_settings={"unitCell": True}),
)

We transform the ase Atoms object into a metatomic system and define the options for the neighbor list.
system = System(
types=torch.from_numpy(atoms.get_atomic_numbers()),
positions=torch.from_numpy(atoms.positions),
cell=torch.from_numpy(atoms.cell.array),
pbc=torch.from_numpy(atoms.pbc),
)
nlo = NeighborListOptions(cutoff=7.0, full_list=False, strict=False)
calculator = NeighborList(nlo, length_unit="Angstrom")
neighbors = calculator.compute(system)
system.add_neighbor_list(nlo, neighbors)
Neighbor lists are stored within metatensor as metatensor.TensorBlock
objects, if you’re curious
neighbors
TensorBlock
samples (6001): ['first_atom', 'second_atom', 'cell_shift_a', 'cell_shift_b', 'cell_shift_c']
components (3): ['xyz']
properties (1): ['distance']
gradients: None
Helper functions for molecular geometry¶
In order to compute the different terms in the Q-TIP4P/f potential, we need to extract some information on the geometry of the water molecule. To keep the model class clean, we define a helper functions that do two things.
First, it computes O-H covalent bond lengths and angle. We use heuristics to identify the covalent bonds as the shortest O-H distances in a simulation. This is necessary when using the model with an external code, that might re-order atoms internally (as is e.g. the case for LAMMPS). The heuristics here may fail in case molecules get too close together, or at very high temperature.
Second, it computes the position of the virtual “M sites”, the position of the O
charge in a TIP4P-like model. We also need distances to compute range-separated
electrostatics, which we obtain manipulating the neighbor list that is pre-attached to
the system. Thanks to the fact we rely on torch autodifferentiation mechanism, the
forces acting on the virtual sites will be automatically split between O and H atoms,
in a way that is consistent with the definition.
Forces acting on the M sites will be automatically split between O and H atoms, in a way that is consistent with the definition.
def get_molecular_geometry(
system: System,
nlo: NeighborListOptions,
m_gamma: torch.Tensor,
m_charge: torch.Tensor,
) -> Tuple[torch.Tensor, torch.Tensor, System, torch.Tensor, torch.Tensor]:
"""Compute bond distances, angles and charge site positions for each molecules."""
neighbors = system.get_neighbor_list(nlo)
species = system.types
# get neighbor idx and vectors as torch tensors
neigh_ij = torch.stack(
(
neighbors.samples.column("first_atom"),
neighbors.samples.column("second_atom"),
),
dim=1,
)
neigh_dij = neighbors.values.squeeze()
# get all OH distances and their sorting order
oh_mask = species[neigh_ij[:, 0]] != species[neigh_ij[:, 1]]
oh_ij = neigh_ij[oh_mask]
oh_dij = neigh_dij[oh_mask]
oh_dist = torch.linalg.vector_norm(oh_dij, dim=1).squeeze()
oh_sort = torch.argsort(oh_dist)
# gets the oxygen indices in the bonds, sorted by distance
oh_oidx = torch.where(species[oh_ij[:, 0]] == 8, oh_ij[:, 0], oh_ij[:, 1])[oh_sort]
# makes sure we always consider bonds in the O->H direction
oh_dij = oh_dij * torch.where(species[oh_ij[:, 0]] == 8, 1.0, -1.0).reshape(-1, 1)
# we assume that the first n_oxygen*2 bonds cover all water molecules.
# if not we throw an error
o_idx = torch.nonzero(species == 8).squeeze()
n_oh = len(o_idx) * 2
oh_oidx_sort = torch.argsort(oh_oidx[:n_oh])
oh_dij_oidx = oh_oidx[oh_oidx_sort] # indices of the O atoms for each dOH
# if each O index appears twice, this should be a vector of zeros
twoo_chk = oh_dij_oidx[::2] - oh_dij_oidx[1::2]
if (twoo_chk != 0).any():
raise RuntimeError("Cannot assign OH bonds to water molecules univocally.")
# finally, we compute O->H1 and O->H2 for each water molecule
oh_dij_sort = oh_dij[oh_sort[:n_oh]][oh_oidx_sort]
doh_1 = oh_dij_sort[::2]
doh_2 = oh_dij_sort[1::2]
oh_dist = torch.concatenate(
[
torch.linalg.vector_norm(doh_1, dim=1),
torch.linalg.vector_norm(doh_2, dim=1),
]
)
if oh_dist.max() > 2.0:
raise ValueError(
"Unphysical O-H bond length. Check that the molecules are entire, and "
"atoms are listed in the expected OHH order."
)
hoh_angles = torch.arccos(
torch.sum(doh_1 * doh_2, dim=1)
/ (oh_dist[: len(doh_1)] * oh_dist[len(doh_2) :])
)
# now we use this information to build the M sites
# we first put the O->H vectors in the same order as the
# positions. This allows us to manipulate all atoms at once later
oh1_vecs = torch.zeros_like(system.positions)
oh1_vecs[oh_dij_oidx[::2]] = doh_1
oh2_vecs = torch.zeros_like(system.positions)
oh2_vecs[oh_dij_oidx[1::2]] = doh_2
# we compute the vectors O->M displacing the O to the M sites
om_displacements = (1 - m_gamma) * 0.5 * (oh1_vecs + oh2_vecs)
# creates a new System with the m-sites
m_system = System(
types=system.types,
positions=system.positions + om_displacements,
cell=system.cell,
pbc=system.pbc,
)
# adjust neighbor lists to point at the m sites rather than O atoms. this assumes
# this won't have atoms cross the cutoff, which is of course only approximately
# true, so one should use a slightly larger-than-usual cutoff nb - this is reshaped
# to match the values in a NL tensorblock
nl = system.get_neighbor_list(nlo)
i_idx = nl.samples.column("first_atom")
j_idx = nl.samples.column("second_atom")
m_nl = TensorBlock(
nl.values
+ (om_displacements[j_idx] - om_displacements[i_idx]).reshape(-1, 3, 1),
nl.samples,
nl.components,
nl.properties,
)
m_system.add_neighbor_list(nlo, m_nl)
# set charges of all atoms
charges = (
torch.where(species == 8, -m_charge, 0.5 * m_charge)
.reshape(-1, 1)
.to(dtype=system.positions.dtype, device=system.positions.device)
)
# Create metadata for the charges TensorBlock
samples = Labels(
"atom", torch.arange(len(system), device=charges.device).reshape(-1, 1)
)
properties = Labels(
"charge", torch.zeros(1, 1, device=charges.device, dtype=torch.int32)
)
data = TensorBlock(
values=charges, samples=samples, components=[], properties=properties
)
tensor = TensorMap(
keys=Labels("_", torch.zeros(1, 1, device=charges.device, dtype=torch.int32)),
blocks=[data],
)
m_system.add_data(name="charge", tensor=tensor)
# intra-molecular distances with M sites (for self-energy removal)
hh_dist = torch.sqrt(((doh_1 - doh_2) ** 2).sum(dim=1))
dmh_1 = doh_1 - om_displacements[oh_dij_oidx[::2]]
dmh_2 = doh_2 - om_displacements[oh_dij_oidx[1::2]]
mh_dist = torch.concatenate(
[
torch.linalg.vector_norm(dmh_1, dim=1),
torch.linalg.vector_norm(dmh_2, dim=1),
]
)
return oh_dist, hoh_angles, m_system, mh_dist, hh_dist
Defining the model¶
Armed with these functions, the definitions of bonded and LJ potentials, and the
torchpme.P3MCalculator class, we can define with relatively small effort the
actual model. We do not hard-code the specific parameters of the Q-TIP4P/f potential
(so that in principle this class can be used for classical TIP4P, and - by setting
m_gamma to one - a three-point water model).
A few points worth noting: (1) As discussed above we define a bare Coulomb potential
(that pretty much computes \(1/r\)) which we need to subtract the molecular “self
electrostatic interaction”; (2) units are expected to be Å for distances, kcal/mol for
energies, and radians for angles; (3) model parameters are registered as buffers;
(4) P3M parameters can also be given, in the format returned by
torchpme.tuning.tune_p3m().
The forward call is a relatively straightforward combination of all the helper
functions defined further up in this file, and should be relatively easy to follow.
class WaterModel(torch.nn.Module):
def __init__(
self,
cutoff: float,
lj_sigma: float,
lj_epsilon: float,
m_gamma: float,
m_charge: float,
oh_bond_eq: float,
oh_bond_k: float,
oh_bond_alpha: float,
hoh_angle_eq: float,
hoh_angle_k: float,
p3m_options: Optional[dict] = None,
):
super().__init__()
if p3m_options is None:
# should give a ~1e-4 relative accuracy on forces
p3m_options = (1.4, {"interpolation_nodes": 5, "mesh_spacing": 1.33}, 0)
p3m_smearing, p3m_parameters, _ = p3m_options
self.p3m_calculator = torchpme.metatensor.P3MCalculator(
potential=torchpme.CoulombPotential(
p3m_smearing, prefactor=torchpme.prefactors.kcalmol_A
),
**p3m_parameters,
)
self.coulomb = torchpme.CoulombPotential()
# We use a half neighborlist and allow to have pairs farther than cutoff
# (`strict=False`) since this is not problematic for PME and may speed up the
# computation of the neighbors.
self.nlo = NeighborListOptions(cutoff=cutoff, full_list=False, strict=False)
self.register_buffer("cutoff", torch.tensor(cutoff))
self.register_buffer("lj_sigma", torch.tensor(lj_sigma))
self.register_buffer("lj_epsilon", torch.tensor(lj_epsilon))
self.register_buffer("m_gamma", torch.tensor(m_gamma))
self.register_buffer("m_charge", torch.tensor(m_charge))
self.register_buffer("oh_bond_eq", torch.tensor(oh_bond_eq))
self.register_buffer("oh_bond_k", torch.tensor(oh_bond_k))
self.register_buffer("oh_bond_alpha", torch.tensor(oh_bond_alpha))
self.register_buffer("hoh_angle_eq", torch.tensor(hoh_angle_eq))
self.register_buffer("hoh_angle_k", torch.tensor(hoh_angle_k))
def requested_neighbor_lists(self):
"""Returns the list of neighbor list options that are needed."""
return [self.nlo]
def _setup_systems(
self,
systems: list[System],
selected_atoms: Optional[Labels] = None,
) -> tuple[System, TensorBlock]:
if len(systems) > 1:
raise ValueError(f"only one system supported, got {len(systems)}")
system_i = 0
system = systems[system_i]
# select only real atoms and discard ghosts
# (this is to work in codes like LAMMPS that provide a neighbor
# list that also includes "domain decomposition" neigbhbors)
if selected_atoms is not None:
current_system_mask = selected_atoms.column("system") == system_i
current_atoms = selected_atoms.column("atom")
current_atoms = current_atoms[current_system_mask].to(torch.long)
types = system.types[current_atoms]
positions = system.positions[current_atoms]
system_clean = System(types, positions, system.cell, system.pbc)
for nlo in system.known_neighbor_lists():
system_clean.add_neighbor_list(nlo, system.get_neighbor_list(nlo))
else:
system_clean = system
return system_clean, system.get_neighbor_list(self.nlo)
def forward(
self,
systems: List[System], # noqa
outputs: Dict[str, ModelOutput], # noqa
selected_atoms: Optional[Labels] = None,
) -> Dict[str, TensorMap]: # noqa
if list(outputs.keys()) != ["energy"]:
raise ValueError(
f"`outputs` keys ({', '.join(outputs.keys())}) contain unsupported "
"keys. Only 'energy' is supported."
)
if outputs["energy"].per_atom:
raise NotImplementedError("per-atom energies are not supported.")
system, neighbors = self._setup_systems(systems, selected_atoms)
# compute non-bonded LJ energy
neighbor_indices = torch.stack(
(
neighbors.samples.column("first_atom"),
neighbors.samples.column("second_atom"),
),
dim=1,
)
species = system.types
oo_mask = (species[neighbor_indices[:, 0]] == 8) & (
species[neighbor_indices[:, 1]] == 8
)
oo_distances = torch.linalg.vector_norm(neighbors.values[oo_mask], dim=1)
energy_lj = lennard_jones_pair(
oo_distances, self.lj_sigma, self.lj_epsilon, self.cutoff
).sum()
d_oh, a_hoh, m_system, mh_dist, hh_dist = get_molecular_geometry(
system, self.nlo, self.m_gamma, self.m_charge
)
# intra-molecular energetics
e_bond = bond_energy(
d_oh, self.oh_bond_k, self.oh_bond_alpha, self.oh_bond_eq
).sum()
e_bend = bend_energy(a_hoh, self.hoh_angle_k, self.hoh_angle_eq).sum()
# now this is the long-range part - computed over the M-site system
# m_system, mh_dist, hh_dist = get_msites(system, self.m_gamma, self.m_charge)
m_neighbors = m_system.get_neighbor_list(self.nlo)
potentials = self.p3m_calculator(m_system, m_neighbors).block(0).values
charges = m_system.get_data("charge").block().values
energy_coulomb = (potentials * charges).sum()
# this is the intra-molecular Coulomb interactions, that must be removed
# to avoid double-counting
energy_self = (
(
self.coulomb.from_dist(mh_dist).sum() * (-self.m_charge)
+ self.coulomb.from_dist(hh_dist).sum() * 0.5 * self.m_charge
)
* 0.5
* self.m_charge
* torchpme.prefactors.kcalmol_A
)
# combines all energy terms
energy_tot = e_bond + e_bend + energy_lj + energy_coulomb - energy_self
# Rename property label to follow metatensor's convention for an atomistic model
samples = Labels(
["system"],
torch.arange(len(systems), device=energy_tot.device).reshape(-1, 1),
)
properties = Labels(["energy"], torch.tensor([[0]], device=energy_tot.device))
block = TensorBlock(
values=torch.sum(energy_tot).reshape(-1, 1),
samples=samples,
components=torch.jit.annotate(List[Labels], []),
properties=properties,
)
return {
"energy": TensorMap(
Labels("_", torch.tensor([[0]], device=energy_tot.device)), [block]
),
}
All this class does is take a System and return its energy (as a
metatensor.TensorMap`).
qtip4pf_parameters = dict(
cutoff=7.0,
lj_sigma=3.1589,
lj_epsilon=0.1852,
m_gamma=0.73612,
m_charge=1.1128,
oh_bond_eq=0.9419,
oh_bond_k=2 * 116.09 * 2.287**2,
oh_bond_alpha=2.287,
hoh_angle_eq=107.4 * torch.pi / 180,
hoh_angle_k=87.85,
)
qtip4pf_model = WaterModel(
**qtip4pf_parameters
# uncomment to override default options
# p3m_options = (1.4, {"interpolation_nodes": 5, "mesh_spacing": 1.33}, 0)
)
We re-initilize the system to ask for gradients
system = System(
types=torch.from_numpy(atoms.get_atomic_numbers()),
positions=torch.from_numpy(atoms.positions).requires_grad_(),
cell=torch.from_numpy(atoms.cell.array).requires_grad_(),
pbc=torch.from_numpy(atoms.pbc),
)
system.add_neighbor_list(nlo, calculator.compute(system))
energy_unit = "kcal/mol"
length_unit = "angstrom"
outputs = {"energy": ModelOutput(quantity="energy", unit=energy_unit, per_atom=False)}
nrg = qtip4pf_model.forward([system], outputs)
nrg["energy"].block(0).values.backward()
print(f"""
Energy is {nrg["energy"].block(0).values[0].item()} kcal/mol
The forces on the first molecule (in kcal/mol/Å) are
{system.positions.grad[:3]}
The stress is
{system.cell.grad}
""")
/home/runner/work/atomistic-cookbook/atomistic-cookbook/examples/water-model/water-model.py:725: DeprecationWarning: `per_atom` is deprecated, please use `sample_kind` instead (Triggered internally at /project/metatomic-torch/src/model.cpp:282.)
if outputs["energy"].per_atom:
Energy is -274.07545584268337 kcal/mol
The forces on the first molecule (in kcal/mol/Å) are
tensor([[-22.5891, -4.0345, 16.9057],
[ 10.1369, 0.0815, -0.2837],
[ 1.1909, 9.2458, -7.3839]], dtype=torch.float64)
The stress is
tensor([[ -1.1619, 15.5917, -14.0268],
[ 18.7409, 5.3058, -16.6880],
[-31.6888, -8.1921, -42.9294]], dtype=torch.float64)
Build and save an AtomisticModel¶
This model can be wrapped into a
metatomic.torch.AtomisticModel class, that provides
useful helpers to specify the capabilities of the model, and to save it as a
torchscript module.
Model options include a definition of its units, and a description of the quantities it can compute.
Note
We need to specify that the model has infinite interaction range because of the presence of a long-range term that means one cannot assume that forces decay to zero beyond the cutoff.
options = ModelEvaluationOptions(
length_unit=length_unit, outputs=outputs, selected_atoms=None
)
model_capabilities = ModelCapabilities(
outputs=outputs,
atomic_types=[1, 8],
interaction_range=torch.inf,
length_unit=length_unit,
supported_devices=["cuda", "cpu"],
dtype="float32",
)
atomistic_model = AtomisticModel(
qtip4pf_model.eval(), ModelMetadata(), model_capabilities
)
atomistic_model.save("qtip4pf-mta.pt")
Other water models¶
The WaterModel class is flexible enough that one can also implement (and export) other 4-point models, or even 3-point models if one sets the m_gamma parameter to one. For instance, we can implement the (classical) SPC/Fw model (Wu et al., JCP (2006))
spcf_parameters = dict(
cutoff=7.0,
lj_sigma=3.16549,
lj_epsilon=0.155425,
m_gamma=1.0,
m_charge=0.82,
oh_bond_eq=1.012,
oh_bond_k=1059.162,
oh_bond_alpha=0.0,
hoh_angle_eq=113.24 * torch.pi / 180,
hoh_angle_k=75.90,
)
spcf_model = WaterModel(**spcf_parameters)
atomistic_model = AtomisticModel(spcf_model.eval(), ModelMetadata(), model_capabilities)
atomistic_model.save("spcfw-mta.pt")
Using the Q-TIP4P/f model¶
The torchscript model can be reused with any simulation software compatible with
the metatomic API. Here we give a couple of examples, designed to demonstrate the
usage more than to provide realistic use cases.
Geometry optimization with ase¶
We begin with an example based on an ase-compatible calculator. To this end,
metatomic provides a compatible
metatomic.torch.MetatomicCalculator wrapper to a model. Note how
the metadata associated with the model are used to convert energy into the units
expected by ase (eV and Å).
atomistic_model = load_atomistic_model("qtip4pf-mta.pt")
mta_calculator = MetatomicCalculator(atomistic_model)
atoms.calc = mta_calculator
nrg = atoms.get_potential_energy()
print(f"""
Energy is {nrg} eV, corresponding to {nrg * 23.060548} kcal/mol
""")
Energy is -11.885025024414062 eV, corresponding to -274.0751900567017 kcal/mol
We then use one of the built-in ase functions to run the structural relaxation.
The relaxation is split into short segments just to be able to visualize the
trajectory.
fmax is the threshold on the maximum force component and the optimization will
stop when the threshold is reached
opt_trj = []
opt_nrg = []
for _ in range(10):
opt_trj.append(atoms.copy())
opt_nrg.append(atoms.get_potential_energy())
opt = LBFGS(atoms, restart="lbfgs_restart.json")
opt.run(fmax=0.001, steps=5)
opt.run(fmax=0.001, steps=10)
nrg_final = atoms.get_potential_energy()
Step Time Energy fmax
LBFGS: 0 08:17:08 -11.885025 3.466987
LBFGS: 1 08:17:08 -12.927923 1.706438
LBFGS: 2 08:17:08 -13.351147 1.339523
LBFGS: 3 08:17:08 -14.188912 1.595493
LBFGS: 4 08:17:09 -14.462056 0.648452
LBFGS: 5 08:17:09 -14.683365 0.598442
Step Time Energy fmax
LBFGS: 0 08:17:09 -14.683365 0.598442
LBFGS: 1 08:17:09 -14.961505 0.829780
LBFGS: 2 08:17:09 -15.255484 0.781364
LBFGS: 3 08:17:09 -15.421952 0.612538
LBFGS: 4 08:17:09 -15.511645 0.412006
LBFGS: 5 08:17:09 -15.616668 0.654147
Step Time Energy fmax
LBFGS: 0 08:17:09 -15.616668 0.654147
LBFGS: 1 08:17:09 -15.758744 0.819039
LBFGS: 2 08:17:09 -15.908655 0.641309
LBFGS: 3 08:17:09 -16.021681 0.450587
LBFGS: 4 08:17:09 -16.081858 0.417818
LBFGS: 5 08:17:09 -16.140636 0.373996
Step Time Energy fmax
LBFGS: 0 08:17:09 -16.140636 0.373996
LBFGS: 1 08:17:09 -16.227831 0.516878
LBFGS: 2 08:17:09 -16.308926 0.410067
LBFGS: 3 08:17:09 -16.372448 0.393186
LBFGS: 4 08:17:09 -16.429533 0.366448
LBFGS: 5 08:17:09 -16.498772 0.375328
Step Time Energy fmax
LBFGS: 0 08:17:09 -16.498772 0.375328
LBFGS: 1 08:17:09 -16.560049 0.312226
LBFGS: 2 08:17:09 -16.603327 0.306848
LBFGS: 3 08:17:09 -16.635597 0.247830
LBFGS: 4 08:17:09 -16.677351 0.289583
LBFGS: 5 08:17:09 -16.740196 0.359899
Step Time Energy fmax
LBFGS: 0 08:17:09 -16.740196 0.359899
LBFGS: 1 08:17:09 -16.812737 0.307706
LBFGS: 2 08:17:09 -16.857498 0.333321
LBFGS: 3 08:17:09 -16.883671 0.346673
LBFGS: 4 08:17:09 -16.913441 0.282538
LBFGS: 5 08:17:09 -16.957270 0.312777
Step Time Energy fmax
LBFGS: 0 08:17:09 -16.957270 0.312777
LBFGS: 1 08:17:09 -17.000168 0.350294
LBFGS: 2 08:17:09 -17.030489 0.246551
LBFGS: 3 08:17:09 -17.052172 0.249773
LBFGS: 4 08:17:09 -17.078173 0.241942
LBFGS: 5 08:17:09 -17.114550 0.312425
Step Time Energy fmax
LBFGS: 0 08:17:09 -17.114550 0.312425
LBFGS: 1 08:17:09 -17.151815 0.254452
LBFGS: 2 08:17:09 -17.178030 0.226609
LBFGS: 3 08:17:09 -17.198738 0.197666
LBFGS: 4 08:17:09 -17.221308 0.175373
LBFGS: 5 08:17:09 -17.245573 0.225201
Step Time Energy fmax
LBFGS: 0 08:17:09 -17.245573 0.225201
LBFGS: 1 08:17:09 -17.264799 0.174612
LBFGS: 2 08:17:09 -17.281357 0.202183
LBFGS: 3 08:17:09 -17.300541 0.193873
LBFGS: 4 08:17:09 -17.328152 0.281321
LBFGS: 5 08:17:09 -17.355932 0.239958
Step Time Energy fmax
LBFGS: 0 08:17:09 -17.355932 0.239958
LBFGS: 1 08:17:09 -17.374861 0.192643
LBFGS: 2 08:17:09 -17.386677 0.163911
LBFGS: 3 08:17:09 -17.401159 0.195095
LBFGS: 4 08:17:09 -17.422842 0.326419
LBFGS: 5 08:17:10 -17.444862 0.218075
LBFGS: 6 08:17:10 -17.460022 0.260483
LBFGS: 7 08:17:10 -17.469721 0.159604
LBFGS: 8 08:17:10 -17.479248 0.189494
LBFGS: 9 08:17:10 -17.495850 0.275882
LBFGS: 10 08:17:10 -17.513508 0.245200
LBFGS: 11 08:17:10 -17.526594 0.143200
LBFGS: 12 08:17:10 -17.534639 0.128328
LBFGS: 13 08:17:10 -17.542475 0.160490
LBFGS: 14 08:17:10 -17.552765 0.178781
LBFGS: 15 08:17:10 -17.563562 0.144455
Use chemiscope to visualize the geometry optimization together with the convergence of the energy to a local minimum.
chemiscope.show(
structures=opt_trj,
properties={
"step": 1 + np.arange(0, len(opt_trj)),
"energy": opt_nrg - nrg_final,
},
mode="default",
settings=chemiscope.quick_settings(
map_settings={
"x": {"property": "step", "scale": "log"},
"y": {"property": "energy", "scale": "log"},
},
structure_settings={
"unitCell": True,
},
trajectory=True,
),
)
Path integral molecular dynamics with i-PI¶
We use i-PI to perform path integral molecular-dynamics simulations of bulk water to include nuclear quntum effects. See this recipe for an introduction to constant-temperature MD using i-PI.
First, the XML input of the i-PI simulation is created using a few utility
functions. This input could also be written to file and used with the command-line
version of i-PI. We use the structure twice to generate a path integral with two
replicas: this is far from converged, see also this recipe if you
have never run path integral simulations before.
data = ase.io.read("data/water_32.xyz")
input_xml = simulation_xml(
structures=[data, data],
forcefield=forcefield_xml(
name="qtip4pf",
mode="direct",
pes="metatomic",
parameters={"model": "qtip4pf-mta.pt", "template": "data/water_32.xyz"},
),
motion=motion_nvt_xml(timestep=0.5 * ase.units.fs),
temperature=300,
prefix="qtip4pf-md",
)
print(input_xml)
<simulation verbosity='quiet' safe_stride='20'>
<ffdirect name='qtip4pf'>
<pes>metatomic</pes>
<parameters>{model: qtip4pf-mta.pt, template: data/water_32.xyz}</parameters>
</ffdirect>
<output prefix='qtip4pf-md'>
<properties stride='2' filename='out'>[ step, time{picosecond}, conserved{electronvolt}, temperature{kelvin}, potential{electronvolt} ]</properties>
<trajectory filename='pos' stride='20' cell_units='angstrom'>positions{angstrom}</trajectory>
<checkpoint stride='200'>
</checkpoint>
</output>
<system>
<beads natoms='96' nbeads='2'>
<q shape='(2, 288)'>
[ 7.09781133e+00, 8.90061005e+00, 1.79410598e+01, 8.70029908e+00, 8.07291001e+00,
1.82755414e+01, 7.55512505e+00, 1.00533430e+01, 1.66069132e+01, 1.87706496e+01,
1.67070687e+01, 6.91639762e-01, 1.91467051e+01, 1.54881953e+01, 2.11649326e+00,
1.77029543e+01, 1.59662960e+01, -5.97153456e-01, 6.06602086e-01, 2.81947138e+00,
1.09528526e+01, -5.42351398e-01, 3.76622417e+00, 9.90405463e+00, 1.49477337e+00,
3.89472555e+00, 1.20262171e+01, 1.51839494e+01, 1.83964838e+01, 8.13905042e+00,
1.36116973e+01, 1.84985290e+01, 9.15950253e+00, 1.63196748e+01, 1.75876811e+01,
9.29745254e+00, 1.07015191e+01, 1.71624927e+01, 1.24721924e+00, 1.20526732e+01,
1.83700277e+01, 1.53823707e+00, 9.85114229e+00, 1.69905276e+01, 2.93285495e+00,
1.53634734e+01, 4.06291117e-01, 2.26956108e+00, 1.54881953e+01, -1.22832198e-01,
4.00243993e+00, 1.68903721e+01, -3.04245906e-01, 1.54579597e+00, 1.54522905e+01,
7.87070931e+00, 1.35304391e+00, 1.49193878e+01, 9.57335255e+00, 1.58736995e+00,
1.45924651e+01, 6.73120446e+00, 2.53412273e+00, 1.19827534e+01, 6.15294827e+00,
1.82887694e+01, 1.34794165e+01, 6.46853253e+00, 1.92544195e+01, 1.22964479e+01,
4.53156325e+00, 1.76689393e+01, 2.57002753e-01, 1.47360843e+01, 5.17407013e+00,
-1.13383568e-02, 1.56847268e+01, 6.77277843e+00, 5.93374004e-01, 1.30485589e+01,
5.60492769e+00, 9.49965323e+00, 4.84336806e+00, 1.16577205e+01, 1.04653033e+01,
6.30412636e+00, 1.18334650e+01, 1.00401149e+01, 3.83803376e+00, 1.30806842e+01,
1.35379980e+01, 4.80368381e+00, 4.56935777e+00, 1.18070088e+01, 4.66195435e+00,
5.03989958e+00, 1.39537377e+01, 3.17662962e+00, 3.88716664e+00, 6.30412636e+00,
1.71039112e+01, 6.16995580e+00, 5.32524822e+00, 1.57622056e+01, 5.26666671e+00,
5.16462150e+00, 1.85079777e+01, 6.42128938e+00, 2.78923576e+00, 2.68341110e+00,
1.54768570e+00, 2.00310969e+00, 1.07336444e+00, 1.19241719e+00, 2.98954673e+00,
2.85159672e+00, 3.34481524e+00, 2.30924533e+00, 9.34658542e+00, 6.08113867e+00,
3.98921185e+00, 8.88738197e+00, 6.65561542e+00, 2.53601246e+00, 9.36548268e+00,
4.24243515e+00, 1.28312404e+01, 1.22662123e+01, 8.48109085e+00, 1.33849301e+01,
1.08753739e+01, 9.58847036e+00, 1.27499822e+01, 1.37817726e+01, 9.40138748e+00,
1.76311448e+01, 6.54790103e+00, 8.37148674e+00, 1.88481284e+01, 7.47575655e+00,
7.40394696e+00, 1.62195193e+01, 5.93940921e+00, 7.49465382e+00, 1.41427103e+01,
8.58502579e+00, 1.20545630e+01, 1.43694775e+01, 8.40928126e+00, 1.38479131e+01,
1.55732330e+01, 7.74409766e+00, 1.13137903e+01, 8.46597304e-01, 1.07733286e+01,
1.55316590e+01, 1.08281307e+00, 9.14627445e+00, 1.48740343e+01, 1.36060281e+00,
1.06788423e+01, 1.72399714e+01, 1.39083843e+00, 7.71386205e+00, 1.02990074e+00,
1.95019736e+00, 5.94696812e+00, 9.46752789e-01, -4.72431531e-01, 7.51733053e+00,
8.99509636e-01, 7.99165179e+00, 4.87927286e+00, 6.73120446e+00, 8.78722649e+00,
4.46731256e+00, 8.36014838e+00, 8.13716070e+00, 6.66317432e+00, 6.60648254e+00,
7.08458325e+00, 1.00495635e+01, 6.78600652e+00, 6.75955035e+00, 1.05824663e+01,
5.08336328e+00, 8.53967236e+00, 1.09830882e+01, 7.23954079e+00, 4.57313722e-01,
1.61382611e+01, 9.81712722e+00, 4.59203449e-01, 1.78616913e+01, 1.03065663e+01,
1.37005144e+00, 1.53105611e+01, 1.10813540e+01, 1.14611890e+01, 4.59203449e-01,
1.54409522e+01, 1.26328192e+01, -6.80301405e-01, 1.61741659e+01, 9.89271627e+00,
-3.77945225e-02, 1.62251885e+01, 1.39121637e+01, 1.51726111e+01, 1.71001317e+01,
1.26970698e+01, 1.47984453e+01, 1.57074036e+01, 1.28747041e+01, 1.48135631e+01,
1.84380578e+01, 3.72465019e+00, 2.21286929e+00, 6.86159556e+00, 2.87238371e+00,
2.64372685e+00, 8.45463469e+00, 5.33091740e+00, 2.98009810e+00, 6.67640240e+00,
1.45017583e+01, 1.24816411e+01, 4.03267555e+00, 1.34983137e+01, 1.22643226e+01,
5.51422084e+00, 1.61817248e+01, 1.26252602e+01, 4.55235024e+00, 5.23265164e+00,
1.39537377e+01, 1.37477576e+01, 4.44652557e+00, 1.52841049e+01, 1.46415980e+01,
4.24999406e+00, 1.23852650e+01, 1.39990911e+01, 4.25755296e+00, 5.27422562e+00,
1.38875973e+01, 5.13627561e+00, 6.10381539e+00, 1.52614282e+01, 5.58414070e+00,
5.04745848e+00, 1.26743931e+01, 6.90127981e+00, 1.88084441e+01, 1.75555557e+01,
7.77433328e+00, 1.79694057e+01, 1.88651359e+01, 6.04523388e+00, 2.01047963e+01,
1.84890804e+01, 9.94562860e+00, 1.26762829e+01, 1.43411316e+01, 8.15983741e+00,
1.33830404e+01, 1.45641193e+01, 1.03084560e+01, 1.33357973e+01, 1.27235260e+01,
6.62726952e+00, 1.27008493e+01, 1.92752065e+00, 7.74220794e+00, 1.38006699e+01,
1.14706376e+00, 6.77655789e+00, 1.13081211e+01, 7.91795247e-01, 1.03084560e+01,
1.73779215e+01, 1.11267074e+01, 1.03424711e+01, 1.81451503e+01, 1.27707692e+01,
8.59636415e+00, 1.74572900e+01, 1.03632581e+01, 7.09781133e+00, 8.90061005e+00,
1.79410598e+01, 8.70029908e+00, 8.07291001e+00, 1.82755414e+01, 7.55512505e+00,
1.00533430e+01, 1.66069132e+01, 1.87706496e+01, 1.67070687e+01, 6.91639762e-01,
1.91467051e+01, 1.54881953e+01, 2.11649326e+00, 1.77029543e+01, 1.59662960e+01,
-5.97153456e-01, 6.06602086e-01, 2.81947138e+00, 1.09528526e+01, -5.42351398e-01,
3.76622417e+00, 9.90405463e+00, 1.49477337e+00, 3.89472555e+00, 1.20262171e+01,
1.51839494e+01, 1.83964838e+01, 8.13905042e+00, 1.36116973e+01, 1.84985290e+01,
9.15950253e+00, 1.63196748e+01, 1.75876811e+01, 9.29745254e+00, 1.07015191e+01,
1.71624927e+01, 1.24721924e+00, 1.20526732e+01, 1.83700277e+01, 1.53823707e+00,
9.85114229e+00, 1.69905276e+01, 2.93285495e+00, 1.53634734e+01, 4.06291117e-01,
2.26956108e+00, 1.54881953e+01, -1.22832198e-01, 4.00243993e+00, 1.68903721e+01,
-3.04245906e-01, 1.54579597e+00, 1.54522905e+01, 7.87070931e+00, 1.35304391e+00,
1.49193878e+01, 9.57335255e+00, 1.58736995e+00, 1.45924651e+01, 6.73120446e+00,
2.53412273e+00, 1.19827534e+01, 6.15294827e+00, 1.82887694e+01, 1.34794165e+01,
6.46853253e+00, 1.92544195e+01, 1.22964479e+01, 4.53156325e+00, 1.76689393e+01,
2.57002753e-01, 1.47360843e+01, 5.17407013e+00, -1.13383568e-02, 1.56847268e+01,
6.77277843e+00, 5.93374004e-01, 1.30485589e+01, 5.60492769e+00, 9.49965323e+00,
4.84336806e+00, 1.16577205e+01, 1.04653033e+01, 6.30412636e+00, 1.18334650e+01,
1.00401149e+01, 3.83803376e+00, 1.30806842e+01, 1.35379980e+01, 4.80368381e+00,
4.56935777e+00, 1.18070088e+01, 4.66195435e+00, 5.03989958e+00, 1.39537377e+01,
3.17662962e+00, 3.88716664e+00, 6.30412636e+00, 1.71039112e+01, 6.16995580e+00,
5.32524822e+00, 1.57622056e+01, 5.26666671e+00, 5.16462150e+00, 1.85079777e+01,
6.42128938e+00, 2.78923576e+00, 2.68341110e+00, 1.54768570e+00, 2.00310969e+00,
1.07336444e+00, 1.19241719e+00, 2.98954673e+00, 2.85159672e+00, 3.34481524e+00,
2.30924533e+00, 9.34658542e+00, 6.08113867e+00, 3.98921185e+00, 8.88738197e+00,
6.65561542e+00, 2.53601246e+00, 9.36548268e+00, 4.24243515e+00, 1.28312404e+01,
1.22662123e+01, 8.48109085e+00, 1.33849301e+01, 1.08753739e+01, 9.58847036e+00,
1.27499822e+01, 1.37817726e+01, 9.40138748e+00, 1.76311448e+01, 6.54790103e+00,
8.37148674e+00, 1.88481284e+01, 7.47575655e+00, 7.40394696e+00, 1.62195193e+01,
5.93940921e+00, 7.49465382e+00, 1.41427103e+01, 8.58502579e+00, 1.20545630e+01,
1.43694775e+01, 8.40928126e+00, 1.38479131e+01, 1.55732330e+01, 7.74409766e+00,
1.13137903e+01, 8.46597304e-01, 1.07733286e+01, 1.55316590e+01, 1.08281307e+00,
9.14627445e+00, 1.48740343e+01, 1.36060281e+00, 1.06788423e+01, 1.72399714e+01,
1.39083843e+00, 7.71386205e+00, 1.02990074e+00, 1.95019736e+00, 5.94696812e+00,
9.46752789e-01, -4.72431531e-01, 7.51733053e+00, 8.99509636e-01, 7.99165179e+00,
4.87927286e+00, 6.73120446e+00, 8.78722649e+00, 4.46731256e+00, 8.36014838e+00,
8.13716070e+00, 6.66317432e+00, 6.60648254e+00, 7.08458325e+00, 1.00495635e+01,
6.78600652e+00, 6.75955035e+00, 1.05824663e+01, 5.08336328e+00, 8.53967236e+00,
1.09830882e+01, 7.23954079e+00, 4.57313722e-01, 1.61382611e+01, 9.81712722e+00,
4.59203449e-01, 1.78616913e+01, 1.03065663e+01, 1.37005144e+00, 1.53105611e+01,
1.10813540e+01, 1.14611890e+01, 4.59203449e-01, 1.54409522e+01, 1.26328192e+01,
-6.80301405e-01, 1.61741659e+01, 9.89271627e+00, -3.77945225e-02, 1.62251885e+01,
1.39121637e+01, 1.51726111e+01, 1.71001317e+01, 1.26970698e+01, 1.47984453e+01,
1.57074036e+01, 1.28747041e+01, 1.48135631e+01, 1.84380578e+01, 3.72465019e+00,
2.21286929e+00, 6.86159556e+00, 2.87238371e+00, 2.64372685e+00, 8.45463469e+00,
5.33091740e+00, 2.98009810e+00, 6.67640240e+00, 1.45017583e+01, 1.24816411e+01,
4.03267555e+00, 1.34983137e+01, 1.22643226e+01, 5.51422084e+00, 1.61817248e+01,
1.26252602e+01, 4.55235024e+00, 5.23265164e+00, 1.39537377e+01, 1.37477576e+01,
4.44652557e+00, 1.52841049e+01, 1.46415980e+01, 4.24999406e+00, 1.23852650e+01,
1.39990911e+01, 4.25755296e+00, 5.27422562e+00, 1.38875973e+01, 5.13627561e+00,
6.10381539e+00, 1.52614282e+01, 5.58414070e+00, 5.04745848e+00, 1.26743931e+01,
6.90127981e+00, 1.88084441e+01, 1.75555557e+01, 7.77433328e+00, 1.79694057e+01,
1.88651359e+01, 6.04523388e+00, 2.01047963e+01, 1.84890804e+01, 9.94562860e+00,
1.26762829e+01, 1.43411316e+01, 8.15983741e+00, 1.33830404e+01, 1.45641193e+01,
1.03084560e+01, 1.33357973e+01, 1.27235260e+01, 6.62726952e+00, 1.27008493e+01,
1.92752065e+00, 7.74220794e+00, 1.38006699e+01, 1.14706376e+00, 6.77655789e+00,
1.13081211e+01, 7.91795247e-01, 1.03084560e+01, 1.73779215e+01, 1.11267074e+01,
1.03424711e+01, 1.81451503e+01, 1.27707692e+01, 8.59636415e+00, 1.74572900e+01,
1.03632581e+01 ]
</q>
<p shape='(2, 288)'>
[ 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00 ]
</p>
<m shape='(96)'>
[ 2.91651223e+04, 1.83736223e+03, 1.83736223e+03, 2.91651223e+04, 1.83736223e+03,
1.83736223e+03, 2.91651223e+04, 1.83736223e+03, 1.83736223e+03, 2.91651223e+04,
1.83736223e+03, 1.83736223e+03, 2.91651223e+04, 1.83736223e+03, 1.83736223e+03,
2.91651223e+04, 1.83736223e+03, 1.83736223e+03, 2.91651223e+04, 1.83736223e+03,
1.83736223e+03, 2.91651223e+04, 1.83736223e+03, 1.83736223e+03, 2.91651223e+04,
1.83736223e+03, 1.83736223e+03, 2.91651223e+04, 1.83736223e+03, 1.83736223e+03,
2.91651223e+04, 1.83736223e+03, 1.83736223e+03, 2.91651223e+04, 1.83736223e+03,
1.83736223e+03, 2.91651223e+04, 1.83736223e+03, 1.83736223e+03, 2.91651223e+04,
1.83736223e+03, 1.83736223e+03, 2.91651223e+04, 1.83736223e+03, 1.83736223e+03,
2.91651223e+04, 1.83736223e+03, 1.83736223e+03, 2.91651223e+04, 1.83736223e+03,
1.83736223e+03, 2.91651223e+04, 1.83736223e+03, 1.83736223e+03, 2.91651223e+04,
1.83736223e+03, 1.83736223e+03, 2.91651223e+04, 1.83736223e+03, 1.83736223e+03,
2.91651223e+04, 1.83736223e+03, 1.83736223e+03, 2.91651223e+04, 1.83736223e+03,
1.83736223e+03, 2.91651223e+04, 1.83736223e+03, 1.83736223e+03, 2.91651223e+04,
1.83736223e+03, 1.83736223e+03, 2.91651223e+04, 1.83736223e+03, 1.83736223e+03,
2.91651223e+04, 1.83736223e+03, 1.83736223e+03, 2.91651223e+04, 1.83736223e+03,
1.83736223e+03, 2.91651223e+04, 1.83736223e+03, 1.83736223e+03, 2.91651223e+04,
1.83736223e+03, 1.83736223e+03, 2.91651223e+04, 1.83736223e+03, 1.83736223e+03,
2.91651223e+04, 1.83736223e+03, 1.83736223e+03, 2.91651223e+04, 1.83736223e+03,
1.83736223e+03 ]
</m>
<names shape='(96)'>
[ O, H, H, O, H,
H, O, H, H, O,
H, H, O, H, H,
O, H, H, O, H,
H, O, H, H, O,
H, H, O, H, H,
O, H, H, O, H,
H, O, H, H, O,
H, H, O, H, H,
O, H, H, O, H,
H, O, H, H, O,
H, H, O, H, H,
O, H, H, O, H,
H, O, H, H, O,
H, H, O, H, H,
O, H, H, O, H,
H, O, H, H, O,
H, H, O, H, H,
O, H, H, O, H,
H ]
</names>
</beads>
<cell shape='(3, 3)'>
[ 1.93885901e+01, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 1.93885901e+01,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 1.93885901e+01 ]
</cell>
<initialize nbeads='2'><velocities mode='thermal' units='ase'> 300 </velocities></initialize><ensemble><temperature units='ase'> 300 </temperature></ensemble>
<forces>
<force forcefield='qtip4pf'> </force>
</forces>
<motion mode="dynamics">
<dynamics mode="nvt">
<timestep units="ase"> 0.04911347394232032 </timestep>
<thermostat mode='svr'>
<tau units='ase'> 0.4911347394232032 </tau>
</thermostat>
</dynamics>
</motion>
</system>
</simulation>
Then, we create an InteractiveSimulation object and run a short simulation (purely
for demonstrative purposes)
sim = InteractiveSimulation(input_xml)
sim.run(400)
The simulation generates output files that can be parsed and visualized from Python
data, info = read_output("qtip4pf-md.out")
trj = read_trajectory("qtip4pf-md.pos_0.xyz")
fig, ax = plt.subplots(1, 1, figsize=(4, 3), constrained_layout=True)
ax.plot(data["time"], data["potential"], "b-", label="potential")
ax.plot(data["time"], data["conserved"] - 4, "k-", label="conserved")
ax.set_xlabel("t / ps")
ax.set_ylabel("energy / ev")
ax.legend()
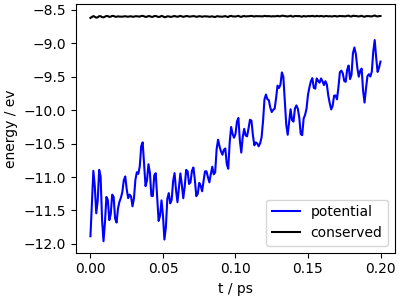
<matplotlib.legend.Legend object at 0x7ff308d3aae0>
chemiscope.show(
structures=trj,
properties={
"time": data["time"][::10],
"potential": data["potential"][::10],
},
mode="default",
settings=chemiscope.quick_settings(
map_settings={
"x": {"property": "time", "scale": "linear"},
"y": {"property": "potential", "scale": "linear"},
},
structure_settings={
"unitCell": True,
},
trajectory=True,
),
)
Molecular dynamics with LAMMPS¶
The metatomic model can also be run with LAMMPS
and used to perform all kinds of atomistic simulations with it.
This only requires defining a pair_metatomic potential, and specifying
the mapping between LAMMPS atom types and those used in the model.
Note also that the metatomic interface takes care of converting the
model units to those used in the LAMMPS file, so it is possible to use
energies in eV even if the model outputs kcal/mol.
with open("data/spcfw.in", "r") as file:
lines = file.readlines()
for line in lines[:7] + lines[16:]:
print(line, end="")
units metal # Angstroms, eV, picoseconds
atom_style atomic
read_data water_32.data
# loads metatomic SPC/Fw model
pair_style metatomic spcfw-mta.pt
pair_coeff * * 1 8
fix 1 all nve
fix 2 all langevin 300 300 1.00 12345
run 200
This specific example runs a short MD trajectory, using a Langevin thermostat. Given that this is a classical MD trajectory, we use the SPC/Fw model that is fitted to reproduce (some) water properties even with a classical description of the nuclei.
We save to geometry to a LAMMPS data file and run the simulation
ase.io.write("water_32.data", atoms, format="lammps-data", masses=True)
run_command("lmp -in data/spcfw.in")
CompletedProcess(args=['lmp', '-in', 'data/spcfw.in'], returncode=0)
Total running time of the script: (0 minutes 26.395 seconds)