Note
Go to the end to download the full example code.
MD using direct-force predictions with PET-MAD¶
- Authors:
Michele Ceriotti @ceriottm, Filippo Bigi @frostedoyster
Evaluating forces as a direct output of a ML model accelerates their evaluation, by up to a factor of 3 in comparison to the traditional approach that evaluates them as derivatives of the interatomic potential. Unfortunately, as discussed e.g. in this paper, doing so means that forces are not conservative, leading to instabilities and artefacts in many modeling tasks, such as constant-energy molecular dynamics. Here we demonstrate the issues associated with direct force predictions, and ways to mitigate them, using the generally-applicable PET-MAD potential. See also this recipe for examples of using PET-MAD for basic tasks such as geometry optimization and conservative MD, and this for an example of how to use direct forces to accelerate training.
# sphinx_gallery_thumbnail_number = 2
If you don’t want to use the conda environment for this recipe, you can get all dependencies installing the upet package:
pip install upet
import linecache
import time
import ase.io
# visualization
import chemiscope
import matplotlib.pyplot as plt
import upet
from atomistic_cookbook_utils import run_command
# i-PI scripting utilities
from ipi.scripting import InteractiveSimulation, read_output, read_trajectory
# metatomic ASE calculator
from metatomic_ase import MetatomicCalculator
from upet.calculator import UPETCalculator
/home/runner/work/atomistic-cookbook/atomistic-cookbook/.nox/pet-mad-nc/lib/python3.12/site-packages/metatomic_ase/_neighbors.py:21: UserWarning: found nvalchemi-toolkit-ops version 0.4.0, this code was only tested with version >=0.3.0,<=0.4.0. If you encounter errors, please update to this version range.
warnings.warn(
Fetch PET-MAD and set up the calculators¶
We use the upet package to download the PET-MAD model. Rather than the
original PET-MAD model, that was trained on PBESol data, we use a model
trained on the MAD-1.5 dataset, that is computed at the r2SCAN level of theory.
See this preprint for details on the
dataset and the training procedure. Here we use the
extra-small (xs) model for speed. For production calculations,
the small (s) model (model="pet-mad-s") is far more accurate and
still very fast.
calculator = UPETCalculator(model="pet-mad-xs", version="1.5.0", device="cpu")
We also save the model as a torchscript file, which is needed for i-PI and LAMMPS integration.
model_path = "pet-mad-xs-v1.5.0.pt"
upet.save_upet(model="pet-mad", size="xs", version="1.5.0", output=model_path)
Warning: You are sending unauthenticated requests to the HF Hub. Please set a HF_TOKEN to enable higher rate limits and faster downloads.
WARNING:huggingface_hub.utils._http:Warning: You are sending unauthenticated requests to the HF Hub. Please set a HF_TOKEN to enable higher rate limits and faster downloads.
/home/runner/work/atomistic-cookbook/atomistic-cookbook/.nox/pet-mad-nc/lib/python3.12/site-packages/metatrain/pet/model.py:1428: UserWarning: the 'non_conservative_forces' output name is deprecated, please update the model to use 'non_conservative_force' instead
return AtomisticModel(self.eval(), metadata, capabilities)
/home/runner/work/atomistic-cookbook/atomistic-cookbook/.nox/pet-mad-nc/lib/python3.12/site-packages/metatrain/pet/model.py:1428: UserWarning: the 'non_conservative_forces' output name is deprecated, please update the model to use 'non_conservative_force' instead
return AtomisticModel(self.eval(), metadata, capabilities)
/home/runner/work/atomistic-cookbook/atomistic-cookbook/.nox/pet-mad-nc/lib/python3.12/site-packages/metatrain/llpr/model.py:999: UserWarning: the 'non_conservative_forces' output name is deprecated, please update the model to use 'non_conservative_force' instead
return AtomisticModel(self.eval(), metadata, self.capabilities)
Non-conservative forces¶
Interatomic potentials are typically used to compute the forces acting on the atoms by differentiating with respect to atomic positions, i.e. if
is the potential for an atomic configuration then the force acting on atom \(i\) is
Even though the early ML-based interatomic potentials followed this route, computing forces directly as a function of the atomic coordinates, as additional heads of the same model that computes \(V\), is computationally more efficient (between 2x and 3x faster). The MetatomicCalculator class allows one to choose between conservative (back-propagated) and non-conservative (direct) force evaluation
structure = ase.io.read("data/bmimcl.xyz")
structure.calc = calculator
energy_c = structure.get_potential_energy()
forces_c = structure.get_forces()
calculator_nc = MetatomicCalculator(
"pet-mad-xs-v1.5.0.pt", device="cpu", non_conservative=True
)
structure.calc = calculator_nc
energy_nc = structure.get_potential_energy()
forces_nc = structure.get_forces()
/home/runner/work/atomistic-cookbook/atomistic-cookbook/.nox/pet-mad-nc/lib/python3.12/site-packages/metatomic/torch/model.py:74: UserWarning: the 'non_conservative_forces' output name is deprecated, please update the model to use 'non_conservative_force' instead
return AtomisticModel(model, model.metadata(), model.capabilities())
print(f"Energy:\n Conservative: {energy_c:.8}\n Non-conserv.: {energy_nc:.8}")
print(
f"Force sample (atom 0):\n Conservative: {forces_c[0]}\n"
+ f" Non-conserv.: {forces_nc[0]}"
)
Energy:
Conservative: -2469.365
Non-conserv.: -2469.365
Force sample (atom 0):
Conservative: [-0.94715989 -1.32085252 -1.65433586]
Non-conserv.: [-0.9724384 -1.25405061 -1.4788754 ]
Energy conservation in NVE molecular dynamics¶
Molecular dynamics simply integrates Hamilton’s equations
to evolve in time the atomic positions. When the forces are the derivatives of a potential energy \(V\), these equations conserve the total energy \(H = V+\sum_i\mathbf{p}_i^2/2m_i\).
Finite time step integrators only conserve energy approximately, but usually stable dynamics implies a high level of conservation, and no long-time drift. Here we demonstrate the impact of direct force prediction on energy conservation, using a short MD trajectory of 1-Butyl-3-methylimidazolium chloride (BMIM-Cl).
Conservative forces¶
First, we run a few steps computing forces as derivatives of the potential.
The MD setup is described in the input-nve.xml file.
This is a rather standard setup, with the key parameters being those
given in the <ffdirect> section.
with open("data/input-nve.xml", "r") as file:
input_nve = file.read()
print(input_nve)
<simulation verbosity="low">
<output prefix="nve-c">
<properties stride="8" filename="out">
[step, time{picosecond}, conserved{electronvolt}, temperature{kelvin},
kinetic_md{electronvolt}, potential{electronvolt},
pot_component(0){electronvolt}
]
</properties>
<trajectory filename="pos" stride="16" format="ase"> positions </trajectory>
<trajectory filename="forces_c" stride="16" format="ase"> forces_component(0) </trajectory>
<checkpoint stride="1000"/>
</output>
<total_steps>32</total_steps>
<prng><seed>12345</seed></prng>
<ffdirect name='cons' pbc="false">
<pes>metatomic</pes>
<parameters>{template:data/bmimcl.xyz,model:pet-mad-xs-v1.5.0.pt,device:cpu,non_conservative:False} </parameters>
</ffdirect>
<system>
<initialize nbeads="1">
<file mode="ase"> data/bmimcl.xyz </file>
<velocities mode="thermal" units="kelvin"> 400.0 </velocities>
</initialize>
<forces>
<force forcefield="cons">
</force>
</forces>
<motion mode="dynamics">
<dynamics mode="nve">
<timestep units="femtosecond"> 0.5 </timestep>
</dynamics>
</motion>
</system>
</simulation>
The simulation can also be run from the command line using
i-pi data/input-nve.xml
but here we run interactively, timing the execution for comparison.
sim = InteractiveSimulation(input_nve)
steps_nve_c = 32
time_nve_c = -time.time()
sim.run(steps_nve_c)
time_nve_c += time.time()
time_nve_c /= steps_nve_c + 1 # there's one extra energy evaluation at the beginning
/home/runner/work/atomistic-cookbook/atomistic-cookbook/.nox/pet-mad-nc/lib/python3.12/site-packages/metatomic/torch/model.py:74: UserWarning: the 'non_conservative_forces' output name is deprecated, please update the model to use 'non_conservative_force' instead
return AtomisticModel(model, model.metadata(), model.capabilities())
@system: Initializing system object
@simulation: Initializing simulation object
@ RANDOM SEED: The seed used in this calculation was 12345
@init_file: Initializing from file data/bmimcl.xyz. Dimension: length, units: automatic, cell_units: automatic
@init_file: Initializing from file data/bmimcl.xyz. Dimension: length, units: automatic, cell_units: automatic
@init_file: Initializing from file data/bmimcl.xyz. Dimension: length, units: automatic, cell_units: automatic
@init_file: Initializing from file data/bmimcl.xyz. Dimension: length, units: automatic, cell_units: automatic
@init_file: Initializing from file data/bmimcl.xyz. Dimension: length, units: automatic, cell_units: automatic
@initializer: Resampling velocities at temperature 400.0 kelvin
@system.bind: Binding the forces
@simulation.run: Average timings at MD step 0. t/step: 1.71607e+00
Non-conservative (direct) forces¶
The PET-MAD model provides direct force predictions, that can be
activated with a non_conservative:True flag. This makes it very
simple to modify the NVE setup:
with open("data/input-nc-nve.xml", "r") as file:
input_nve = file.read()
print(input_nve[574:764])
<ffdirect name='nocons' pbc="false">
<pes>metatomic</pes>
<parameters>{template:data/bmimcl.xyz,model:pet-mad-xs-v1.5.0.pt,device:cpu,non_conservative:True} </parameters>
</ffdirec
We run this example for longer (it is faster!) and time it for comparison
sim = InteractiveSimulation(input_nve)
steps_nve_nc = 128
time_nve_nc = -time.time()
sim.run(steps_nve_nc)
time_nve_nc += time.time()
time_nve_nc /= steps_nve_nc + 1
/home/runner/work/atomistic-cookbook/atomistic-cookbook/.nox/pet-mad-nc/lib/python3.12/site-packages/metatomic/torch/model.py:74: UserWarning: the 'non_conservative_forces' output name is deprecated, please update the model to use 'non_conservative_force' instead
return AtomisticModel(model, model.metadata(), model.capabilities())
@system: Initializing system object
@simulation: Initializing simulation object
@ RANDOM SEED: The seed used in this calculation was 12345
@init_file: Initializing from file data/bmimcl.xyz. Dimension: length, units: automatic, cell_units: automatic
@init_file: Initializing from file data/bmimcl.xyz. Dimension: length, units: automatic, cell_units: automatic
@init_file: Initializing from file data/bmimcl.xyz. Dimension: length, units: automatic, cell_units: automatic
@init_file: Initializing from file data/bmimcl.xyz. Dimension: length, units: automatic, cell_units: automatic
@init_file: Initializing from file data/bmimcl.xyz. Dimension: length, units: automatic, cell_units: automatic
@initializer: Resampling velocities at temperature 400.0 kelvin
@system.bind: Binding the forces
/home/runner/work/atomistic-cookbook/atomistic-cookbook/.nox/pet-mad-nc/lib/python3.12/site-packages/torch/nn/modules/module.py:1787: UserWarning: the 'non_conservative_forces' output name is deprecated, please update the engine to use 'non_conservative_force' instead
return forward_call(*args, **kwargs)
@simulation.run: Average timings at MD step 0. t/step: 1.47003e+00
@open_backup: Backup performed: RESTART -> #RESTART#0#
The simulation generates output files that can be parsed and visualized from Python.
data_c, info = read_output("nve-c.out")
data_nc, info = read_output("nve-nc.out")
There is a large drift of the onserved quantity, that is also associated in a rapid increase of the potential energy, which indicates that the lack of conservative behavior distorts the sampled ensemble (and in fact, would lead to loss of structural integrity in a longer run).
fig, ax = plt.subplots(1, 1, figsize=(4, 3), constrained_layout=True)
ax.set_facecolor("white")
ax.plot(data_c["time"], data_c["potential"], "b*", label=r"$V$ (cons.)")
ax.plot(data_c["time"], data_c["conserved"] - 20, "k*", label=r"$H$ (cons.)")
ax.plot(data_nc["time"], data_nc["potential"], "b--", label=r"$V$ (direct)")
ax.plot(data_nc["time"], data_nc["conserved"] - 20, "k--", label=r"$H$ (direct)")
ax.set_xlabel("t / ps")
ax.set_ylabel("energy / ev")
ax.legend()
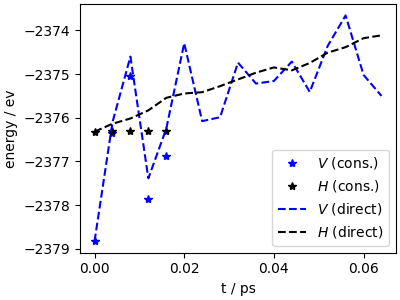
<matplotlib.legend.Legend object at 0x7f2bfbe2e240>
Energy conservation at low-cost with multiple time stepping¶
Given that PET-MAD provides both direct and conservative forces, it is possible to implement a simulation strategy that achieves a high degree of energy conservation at a cost that is close to that of direct-force MD. This relies on the multiple time stepping (MTS) idea, which is discussed and demonstrated in this recipe.
The key idea is to perform several steps of MD using the short timestep needed to follow atomic motion, and a “cheap” force evaluator \(\mathbf{f}_{\mathrm{fast}}\), and then apply a correction \(\mathbf{f}_{\mathrm{slow}}\) every \(M\) of such steps. In this case, the fast forces are the direct predictions, and the slow ones the difference between conservative and direct forces.
This simulation setup can be realized readily in i-PI.
with open("data/input-nc-nve-mts.xml", "r") as file:
input_nve_mts = file.read()
First, one can define two forcefields that compute conservative and direct forces
print(input_nve_mts[704:1082])
<ffdirect name='cons' pbc="false">
<pes>metatomic</pes>
<parameters>{template:data/bmimcl.xyz,model:pet-mad-xs-v1.5.0.pt,device:cpu,non_conservative:False} </parameters>
</ffdirect>
<ffdirect name='nocons' pbc="false">
<pes>metatomic</pes>
<parameters>{template:data/bmimcl.xyz,model:pet-mad-xs-v1.5.0.pt,device:cpu,non_conservative:True} </parameters>
</ffdir
… then, use them in the definition of the systems, specifying how to weight them at each MTS level
print(input_nve_mts[1258:1482])
ize>
<forces>
<force forcefield="cons">
<mts_weights>[1,0]</mts_weights>
</force>
<force forcefield="nocons">
<mts_weights>[-1,1]</mts_weights>
</force>
</force
… and finally request the appropriate MTS discretization in the integrator: this specifies that the inner loop should be executed 8 times, and the outer loop (which has an overall time step of 4 fs) once per step
print(input_nve_mts[1480:1655])
ces>
<motion mode="dynamics">
<dynamics mode="nve">
<timestep units="femtosecond"> 4 </timestep>
<nmts>[1,8]</nmts>
</dynamics>
</mot
All of this happens behind the scenes, and the simulation is just run as for the simpler MD cases. Note also that it is possible to combine this with thermostatted or NPT dynamics, in a completely seamless manner
sim = InteractiveSimulation(input_nve_mts)
nmts = 8
steps_nve_mts = steps_nve_nc // nmts
time_nve_mts = -time.time()
sim.run(steps_nve_mts)
time_nve_mts += time.time()
time_nve_mts /= steps_nve_mts * nmts + 1
/home/runner/work/atomistic-cookbook/atomistic-cookbook/.nox/pet-mad-nc/lib/python3.12/site-packages/metatomic/torch/model.py:74: UserWarning: the 'non_conservative_forces' output name is deprecated, please update the model to use 'non_conservative_force' instead
return AtomisticModel(model, model.metadata(), model.capabilities())
/home/runner/work/atomistic-cookbook/atomistic-cookbook/.nox/pet-mad-nc/lib/python3.12/site-packages/metatomic/torch/model.py:74: UserWarning: the 'non_conservative_forces' output name is deprecated, please update the model to use 'non_conservative_force' instead
return AtomisticModel(model, model.metadata(), model.capabilities())
@system: Initializing system object
@simulation: Initializing simulation object
@ RANDOM SEED: The seed used in this calculation was 12345
@init_file: Initializing from file data/bmimcl.xyz. Dimension: length, units: automatic, cell_units: automatic
@init_file: Initializing from file data/bmimcl.xyz. Dimension: length, units: automatic, cell_units: automatic
@init_file: Initializing from file data/bmimcl.xyz. Dimension: length, units: automatic, cell_units: automatic
@init_file: Initializing from file data/bmimcl.xyz. Dimension: length, units: automatic, cell_units: automatic
@init_file: Initializing from file data/bmimcl.xyz. Dimension: length, units: automatic, cell_units: automatic
@initializer: Resampling velocities at temperature 400.0 kelvin
@system.bind: Binding the forces
/home/runner/work/atomistic-cookbook/atomistic-cookbook/.nox/pet-mad-nc/lib/python3.12/site-packages/torch/nn/modules/module.py:1787: UserWarning: the 'non_conservative_forces' output name is deprecated, please update the engine to use 'non_conservative_force' instead
return forward_call(*args, **kwargs)
@simulation.run: Average timings at MD step 0. t/step: 4.26283e+00
@open_backup: Backup performed: RESTART -> #RESTART#1#
The MTS calculation recovers most of the speedup of direct forces
print(f"""
Time per 0.5fs step:
Conservative forces: {time_nve_c:.4f} s/step
Direct forces: {time_nve_nc:.4f} s/step
MTS (M=8): {time_nve_mts:.4f} s/step
""")
Time per 0.5fs step:
Conservative forces: 0.4104 s/step
Direct forces: 0.2151 s/step
MTS (M=8): 0.2346 s/step
… and the energy conservation is on par with the conservative trajectory (although the actual trajectories would deviate from each other due to accumulation of small errors, but the overall sampling is reliable on a long timescale).
data_mts, info = read_output("nve-nc-mts.out")
trj_mts = read_trajectory("nve-nc-mts.pos_0.extxyz")
force_c_mts = read_trajectory("nve-nc-mts.forces_c.extxyz")
force_nc_mts = read_trajectory("nve-nc-mts.forces_nc.extxyz")
#
fig, ax = plt.subplots(1, 1, figsize=(4, 3), constrained_layout=True)
ax.set_facecolor("white")
ax.plot(data_c["time"], data_c["potential"], "b*", label=r"$V$ (cons.)")
ax.plot(data_nc["time"], data_nc["potential"], "b--", label=r"$V$ (direct)")
ax.plot(data_mts["time"], data_mts["potential"], "b-", label=r"$V$ (MTS)")
ax.plot(data_c["time"], data_c["conserved"] - 20, "k*", label=r"$H$ (cons.)")
ax.plot(data_nc["time"], data_nc["conserved"] - 20, "k--", label=r"$H$ (direct)")
ax.plot(data_mts["time"], data_mts["conserved"] - 20, "k-", label=r"$H$ (MTS)")
ax.set_xlabel("t / ps")
ax.set_ylabel("energy / ev")
ax.legend(ncols=2)
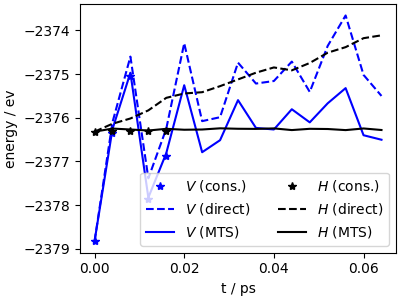
<matplotlib.legend.Legend object at 0x7f2bf0909580>
i-PI prints out both force components for diagnostics, which we can visualize along the (short) trajectory. The conservative forces are shown in atom colors, and the direct predictions in red. One sees clearly that the two predictions are quite close, so the correction is small and can be successfully applied with a large MTS stride.
cs_forces_c = chemiscope.ase_vectors_to_arrows(
force_c_mts, "forces_component", scale=1.0
)
cs_forces_nc = chemiscope.ase_vectors_to_arrows(
force_nc_mts, "forces_component", scale=1.0
)
cs_forces_nc["parameters"]["global"]["color"] = "#aa0000"
chemiscope.show(
trj_mts,
mode="default",
properties={
"time": data_mts["time"][::2],
"conserved": data_mts["conserved"][::2],
"potential": data_mts["potential"][::2],
},
shapes={
"forces_c": cs_forces_c,
"forces_nc": cs_forces_nc,
},
settings=chemiscope.quick_settings(
x="time",
y="potential",
structure_settings={"unitCell": True, "shape": ["forces_c", "forces_nc"]},
trajectory=True,
),
meta={
"name": "MTS direct-forces MD for BMIM-Cl",
"description": "Initial configuration kindly provided "
+ " by Moritz Schaefer and Fabian Zills",
},
)

/home/runner/work/atomistic-cookbook/atomistic-cookbook/examples/pet-mad-nc/pet-mad-nc.py:335: UserWarning: `meta` argument is deprecated, use `metadata` instead
chemiscope.show(
LAMMPS implementation¶
The speedup of the MTS approach with direct forces can also be exploited in LAMMPS. We only show minimal examples running for a few steps to keep the execution time short, but the same approach can be used for realistic simulations. Keep in mind that in order to accelerate the simulation, you should change the cpu device to cuda in the LAMMPS input file when running on a GPU system.
We first launch conservative and non-conservative trajectories for reference. These use the metatomic interface to LAMMPS (which requires a custom LAMMPS build, available through the metatensor conda forge). See also the metatomic documentation for installation instructions.
print(linecache.getline("data/lammps-c.in", 12), end="")
time_lammps_c = -time.time()
run_command("lmp -in data/lammps-c.in")
time_lammps_c += time.time()
pair_style metatomic pet-mad-xs-v1.5.0.pt device cpu
In order to get the non-conservative forces, we just need to
specify the non_conservative on flag in the LAMMPS input file.
print(linecache.getline("data/lammps-nc.in", 12), end="")
time_lammps_nc = -time.time()
run_command("lmp -in data/lammps-nc.in")
time_lammps_nc += time.time()
pair_style metatomic pet-mad-xs-v1.5.0.pt device cpu non_conservative on
The multiple time stepping integrator can be implemented in lammps
using a pair_style hybrid/overlay, providing multiple
metatomic_X pair styles - one for the fast (non-conservative) forces, and two
for the slow correction (conservative minus non-conservative).
Note that you can also use pair_style hybrid/scaled, which however
is affected by a bug at the
time of writing, which prevents it from working correctly with the GPU build
of LAMMPS.
for lineno in [12, 13, 14, 15, 17, 18, 19, 24, 27]:
print(linecache.getline("data/lammps-respa.in", lineno), end="")
time_lammps_mts = -time.time()
run_command("lmp -in data/lammps-respa.in")
time_lammps_mts += time.time()
pair_style hybrid/overlay &
metatomic_1 pet-mad-xs-v1.5.0.pt device cpu non_conservative on scale 1.0 &
metatomic_2 pet-mad-xs-v1.5.0.pt device cpu non_conservative on scale -1.0 &
metatomic_3 pet-mad-xs-v1.5.0.pt device cpu non_conservative off scale 1.0
pair_coeff * * metatomic_1 6 17 1 7
pair_coeff * * metatomic_2 6 17 1 7
pair_coeff * * metatomic_3 6 17 1 7
run_style respa 2 8 hybrid 1 2 2
neigh_modify one 5000
The timings for the three LAMMPS simulations are as follows. Note that while i-PI reuses the fast force for the correction in the outer loop, with the current implementation LAMMPS requires a separate pair style, which reduces the MTS speedup slightly.
print(f"""
Time per 0.5fs step in LAMMPS:
Conservative forces: {time_lammps_c / 16:.4f} s/step
Direct forces: {time_lammps_nc / 16:.4f} s/step
MTS (M=8): {time_lammps_mts / 16:.4f} s/step
""")
Time per 0.5fs step in LAMMPS:
Conservative forces: 0.8084 s/step
Direct forces: 0.5157 s/step
MTS (M=8): 0.9651 s/step
Running LAMMPS on GPUs with KOKKOS¶
If you have a GPU available, you can achieve a dramatic speedup
by running the metatomic model on the GPU, which you can achieve
by setting device cuda for the metatomic pair style in the LAMMPS input files.
The MD integration will however still be run on the CPU, which can become the
bottleneck - especially because atomic positions need to be transferred to the GPU
at each call. LAMMPS can also be run directly on the GPU using the KOKKOS package,
see the installation instructions for
the kokkos-enabled version.
In order to enable the KOKKOS execution, you then have to use additional command-line
arguments when running LAMMPS, e.g.
lmp -k on g <NGPUS> -pk kokkos newton on neigh half -sf kk.
The commands to execute the LAMMPS simulation examples with Kokkos enabled, using
conservative, non-conservative, and MTS force evaluations, are
lmp -k on g 1 -pk kokkos newton on neigh half -sf kk -in data/lammps-c.in
lmp -k on g 1 -pk kokkos newton on neigh half -sf kk -in data/lammps-nc.in
lmp -k on g 1 -pk kokkos newton on neigh half -sf kk -in data/lammps-respa.in
Total running time of the script: (2 minutes 0.506 seconds)