Note
Go to the end to download the full example code.
Thermal conductivity from the Boltzmann transport equation¶
- Authors:
Giuseppe Barbalinardo @gbarbalinardo
This recipe shows how to compute lattice thermal conductivity \(\kappa\) by solving the phonon Boltzmann transport equation (BTE) with kALDo and the PET-MAD universal machine-learning potential via the UPET calculator.
The workflow builds on two companion recipes:
Geometry relaxation with PET-MAD — structure relaxation with a universal MLIP.
Phonon dispersions with PET-MAD — harmonic phonon properties from second-order force constants.
Here we go one step further: we also compute third-order (anharmonic) force constants \(\Phi^{(3)}\), which describe phonon–phonon scattering, and solve the BTE to obtain the thermal conductivity tensor. A comprehensive study of thermal conductivity with universal MLIPs is presented in Barbalinardo et al. (2026). The theory and implementation of kALDo are described in Barbalinardo et al., J. Appl. Phys. 128, 135104 (2020).
We use silicon (diamond) as a test system. The experimental thermal conductivity of natural Si at 300 K is ~150 W/(m·K).
Note
The supercell and k-point grids used here are deliberately small so the recipe runs in CI in a few minutes. See the convergence discussion at the end for production-quality settings.
# sphinx_gallery_thumbnail_number = 7
Setup¶
We use the extra-small (XS) variant of
PET-MAD v1.5.0. For production
calculations, the S model (pet-mad-s) is recommended.
import matplotlib.pyplot as plt
import numpy as np
import chemiscope
from ase.build import bulk
from ase.constraints import FixSymmetry
from ase.filters import StrainFilter
from ase.optimize import BFGS
import kaldo.controllers.plotter as plotter
from kaldo.conductivity import Conductivity
from kaldo.forceconstants import ForceConstants
from kaldo.phonons import Phonons
from upet.calculator import UPETCalculator
plt.rcParams["figure.autolayout"] = True
DEVICE = "cpu"
calc = UPETCalculator(
model="pet-mad-xs",
device=DEVICE,
dtype="float32",
version="1.5.0",
)
/home/runner/work/atomistic-cookbook/atomistic-cookbook/.nox/thermal-conductivity-bte/lib/python3.12/site-packages/metatomic_ase/_neighbors.py:21: UserWarning: found nvalchemi-toolkit-ops version 0.4.0, this code was only tested with version >=0.3.0,<=0.4.0. If you encounter errors, please update to this version range.
warnings.warn(
Warning: You are sending unauthenticated requests to the HF Hub. Please set a HF_TOKEN to enable higher rate limits and faster downloads.
WARNING:huggingface_hub.utils._http:Warning: You are sending unauthenticated requests to the HF Hub. Please set a HF_TOKEN to enable higher rate limits and faster downloads.
Structure relaxation¶
We build a 2-atom Si primitive cell (diamond) and relax the lattice
parameter while preserving symmetry.
FixSymmetry prevents spurious symmetry-breaking that can arise with
unconstrained models, and StrainFilter allows the cell shape to relax
at zero internal stress (see also the geometry relaxation recipe).
atoms = bulk("Si", "diamond", a=5.43)
atoms.calc = calc
atoms.set_constraint(FixSymmetry(atoms))
sf = StrainFilter(atoms)
opt = BFGS(sf, logfile=None)
opt.run(fmax=1e-4)
atoms.set_constraint(None)
a_opt = atoms.cell.cellpar()[0]
print(f"Optimized lattice parameter: {a_opt:.3f} Å")
Optimized lattice parameter: 3.842 Å
Force constants¶
Thermal transport requires two sets of interatomic force constants (IFCs), both computed here by finite differences on a 3×3×3 supercell:
Second-order IFCs \(\Phi^{(2)}\) (harmonic): determine phonon frequencies and group velocities — the ingredients for ballistic (non-interacting) phonon transport. These are the same force constants used in the phonon-dispersion recipe.
Third-order IFCs \(\Phi^{(3)}\) (anharmonic): describe three-phonon scattering processes that limit the phonon mean free path and thus govern diffusive thermal transport.
In production, the second- and third-order supercells can be chosen independently; the third-order calculation is far more expensive because the number of independent displacements scales as \(O(N^2)\) rather than \(O(N)\).
supercell = np.array([3, 3, 3])
chemiscope.show(
[atoms.repeat(supercell)],
mode="structure",
settings=chemiscope.quick_settings(periodic=True),
)

forceconstants = ForceConstants(
atoms=atoms,
supercell=supercell,
third_supercell=supercell,
folder="fd_si/",
is_acoustic_sum=True,
)
forceconstants.second.calculate(calc, delta_shift=3e-2)
forceconstants.third.calculate(calc, delta_shift=3e-2)
2026-07-19 08:17:55,576 - kaldo - INFO - Second order not found. Calculating.
INFO:kaldo:Second order not found. Calculating.
2026-07-19 08:17:55,576 - kaldo - INFO - Calculating second order potential derivatives, finite difference displacement: 3.000e-02 angstrom
INFO:kaldo:Calculating second order potential derivatives, finite difference displacement: 3.000e-02 angstrom
2026-07-19 08:17:55,997 - kaldo - INFO - Symmetry of Dynamical Matrix 0.0028475963821004936
INFO:kaldo:Symmetry of Dynamical Matrix 0.0028475963821004936
2026-07-19 08:17:56,002 - kaldo - INFO - Space group: Fd-3m (#227), 48 unit-cell ops, 48 compatible with supercell shape (3, 3, 3)
INFO:kaldo:Space group: Fd-3m (#227), 48 unit-cell ops, 48 compatible with supercell shape (3, 3, 3)
2026-07-19 08:17:56,006 - kaldo - INFO - Symmetrized order-2 force constants; max change 7.66e-02 (pass symmetrize=False to disable).
INFO:kaldo:Symmetrized order-2 force constants; max change 7.66e-02 (pass symmetrize=False to disable).
2026-07-19 08:17:56,007 - kaldo - INFO - fd_si/second stored
INFO:kaldo:fd_si/second stored
2026-07-19 08:17:56,042 - kaldo - INFO - error sum rule: -1.1866601786144487e-05
INFO:kaldo:error sum rule: -1.1866601786144487e-05
2026-07-19 08:17:56,044 - kaldo - INFO - Third order not found. Calculating.
INFO:kaldo:Third order not found. Calculating.
2026-07-19 08:17:56,044 - kaldo - INFO - Calculating third order potential derivatives, finite difference displacement: 3.000e-02 angstrom
INFO:kaldo:Calculating third order potential derivatives, finite difference displacement: 3.000e-02 angstrom
2026-07-19 08:20:09,363 - kaldo - INFO - Completed atom 1: 50% done
INFO:kaldo:Completed atom 1: 50% done
2026-07-19 08:20:09,364 - kaldo - INFO - Completed atom 0: 100% done
INFO:kaldo:Completed atom 0: 100% done
2026-07-19 08:20:09,364 - kaldo - INFO - total forces to calculate third : 972
INFO:kaldo:total forces to calculate third : 972
2026-07-19 08:20:09,364 - kaldo - INFO - forces calculated : 972
INFO:kaldo:forces calculated : 972
2026-07-19 08:20:09,364 - kaldo - INFO - forces skipped (outside distance threshold) : 0
INFO:kaldo:forces skipped (outside distance threshold) : 0
2026-07-19 08:20:09,399 - kaldo - INFO - Space group: Fd-3m (#227), 48 unit-cell ops, 48 compatible with supercell shape (3, 3, 3)
INFO:kaldo:Space group: Fd-3m (#227), 48 unit-cell ops, 48 compatible with supercell shape (3, 3, 3)
2026-07-19 08:20:15,418 - kaldo - INFO - Symmetrized order-3 force constants; max change 6.04e-01 (pass symmetrize=False to disable).
INFO:kaldo:Symmetrized order-3 force constants; max change 6.04e-01 (pass symmetrize=False to disable).
Harmonic phonon properties¶
We create a Phonons object on a 5×5×5 k-point mesh at 300 K and
inspect the harmonic (second-order) properties: the phonon band structure,
density of states, and mode heat capacities.
kpts = np.array([5, 5, 5])
temperature = 300 # K
phonons = Phonons(
forceconstants=forceconstants,
kpts=kpts,
is_classic=False,
temperature=temperature,
folder="ald_si/",
storage="memory",
is_unfolding=True,
)
2026-07-19 08:20:15,477 - kaldo - INFO - Using unfolding.
INFO:kaldo:Using unfolding.
The phonon dispersion shows the frequency of each phonon mode as a function of wave vector along high-symmetry directions. Silicon has 6 branches (3 acoustic + 3 optical) because the primitive cell contains 2 atoms.
plotter.plot_dispersion(phonons)
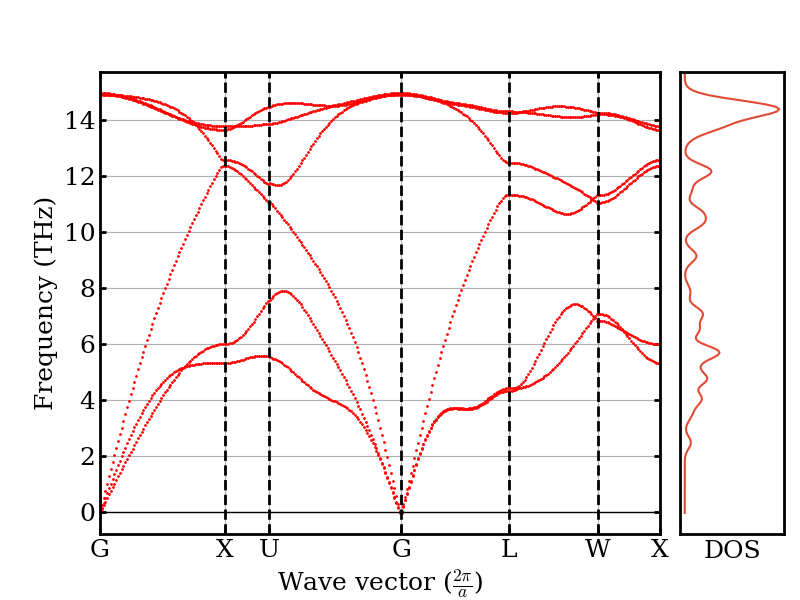
/home/runner/work/atomistic-cookbook/atomistic-cookbook/.nox/thermal-conductivity-bte/lib/python3.12/site-packages/kaldo/controllers/plotter.py:595: UserWarning: This figure includes Axes that are not compatible with tight_layout, so results might be incorrect.
plt.savefig(folder + '/dispersion_dos.png', dpi=300, bbox_inches='tight')
The phonon density of states (DOS) gives the distribution of phonon frequencies. Peaks correspond to flat regions of the dispersion (van Hove singularities).
plotter.plot_dos(phonons)
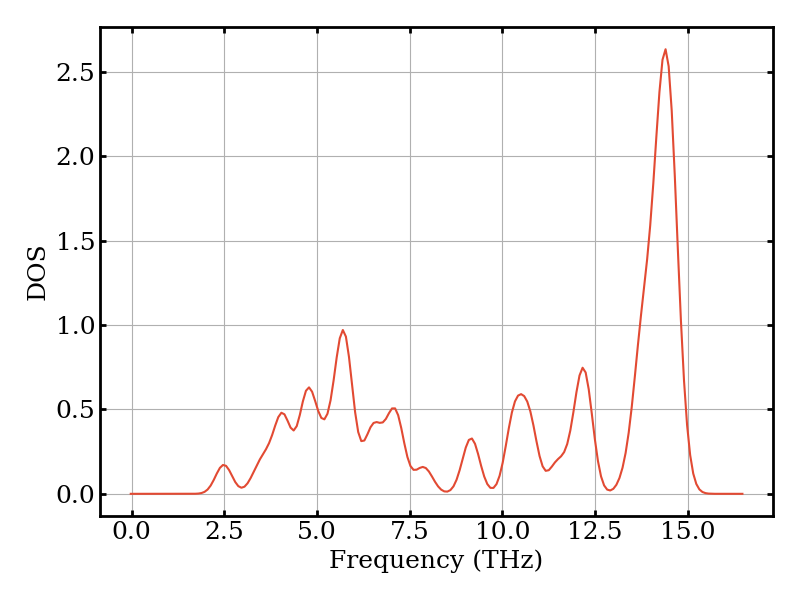
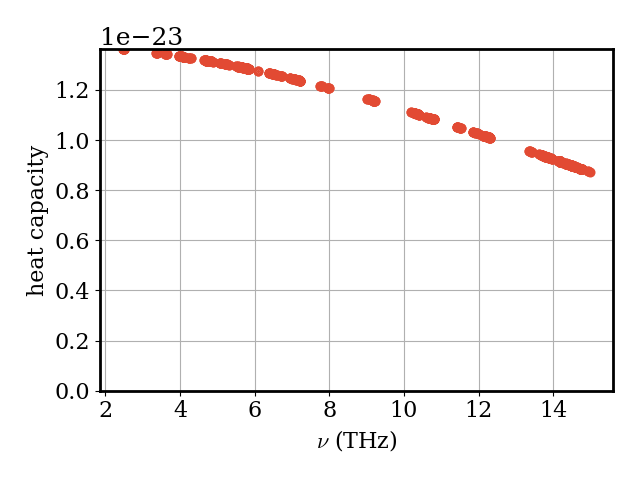
The mode heat capacity \(C_\nu\) tells us how much each phonon mode \(\nu\) contributes to the total heat capacity at this temperature.
plotter.plot_vs_frequency(phonons, phonons.heat_capacity, "heat capacity")
Anharmonic properties and thermal conductivity¶
To obtain the lattice thermal conductivity we need the phonon
lifetimes (inverse bandwidths), which arise from three-phonon
scattering encoded in \(\Phi^{(3)}\). We solve the linearised BTE
using the direct-inversion method (method='inverse'); kALDo also
supports the relaxation-time approximation ('rta') and
self-consistent iteration ('sc').
conductivity = Conductivity(phonons=phonons, method="inverse", storage="memory")
The phonon linewidths (scattering rates) \(\Gamma_\nu\) quantify how strongly each mode is scattered by anharmonic interactions. Modes with larger linewidths have shorter lifetimes and carry less heat.
plotter.plot_vs_frequency(phonons, phonons.bandwidth, "bandwidth")
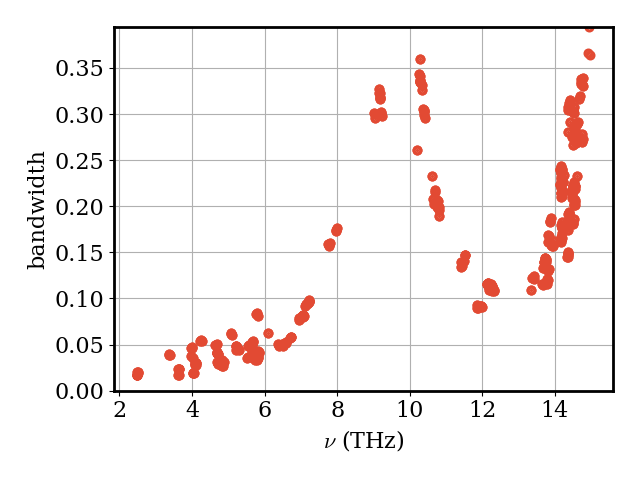
2026-07-19 08:20:18,029 - kaldo - INFO - _ps_gamma_and_gamma_tensor not found.
INFO:kaldo:_ps_gamma_and_gamma_tensor not found.
2026-07-19 08:20:18,765 - kaldo - INFO - Projection started
INFO:kaldo:Projection started
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 1/125 (index 37)
INFO:kaldo:Completed IBZ k-point 1/125 (index 37)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 2/125 (index 72)
INFO:kaldo:Completed IBZ k-point 2/125 (index 72)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 3/125 (index 3)
INFO:kaldo:Completed IBZ k-point 3/125 (index 3)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 4/125 (index 69)
INFO:kaldo:Completed IBZ k-point 4/125 (index 69)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 5/125 (index 101)
INFO:kaldo:Completed IBZ k-point 5/125 (index 101)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 6/125 (index 55)
INFO:kaldo:Completed IBZ k-point 6/125 (index 55)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 7/125 (index 75)
INFO:kaldo:Completed IBZ k-point 7/125 (index 75)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 8/125 (index 59)
INFO:kaldo:Completed IBZ k-point 8/125 (index 59)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 9/125 (index 52)
INFO:kaldo:Completed IBZ k-point 9/125 (index 52)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 10/125 (index 120)
INFO:kaldo:Completed IBZ k-point 10/125 (index 120)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 11/125 (index 89)
INFO:kaldo:Completed IBZ k-point 11/125 (index 89)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 12/125 (index 119)
INFO:kaldo:Completed IBZ k-point 12/125 (index 119)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 13/125 (index 41)
INFO:kaldo:Completed IBZ k-point 13/125 (index 41)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 14/125 (index 53)
INFO:kaldo:Completed IBZ k-point 14/125 (index 53)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 15/125 (index 107)
INFO:kaldo:Completed IBZ k-point 15/125 (index 107)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 16/125 (index 31)
INFO:kaldo:Completed IBZ k-point 16/125 (index 31)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 17/125 (index 51)
INFO:kaldo:Completed IBZ k-point 17/125 (index 51)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 18/125 (index 34)
INFO:kaldo:Completed IBZ k-point 18/125 (index 34)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 19/125 (index 99)
INFO:kaldo:Completed IBZ k-point 19/125 (index 99)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 20/125 (index 25)
INFO:kaldo:Completed IBZ k-point 20/125 (index 25)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 21/125 (index 43)
INFO:kaldo:Completed IBZ k-point 21/125 (index 43)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 22/125 (index 5)
INFO:kaldo:Completed IBZ k-point 22/125 (index 5)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 23/125 (index 45)
INFO:kaldo:Completed IBZ k-point 23/125 (index 45)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 24/125 (index 32)
INFO:kaldo:Completed IBZ k-point 24/125 (index 32)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 25/125 (index 77)
INFO:kaldo:Completed IBZ k-point 25/125 (index 77)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 26/125 (index 81)
INFO:kaldo:Completed IBZ k-point 26/125 (index 81)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 27/125 (index 85)
INFO:kaldo:Completed IBZ k-point 27/125 (index 85)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 28/125 (index 46)
INFO:kaldo:Completed IBZ k-point 28/125 (index 46)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 29/125 (index 16)
INFO:kaldo:Completed IBZ k-point 29/125 (index 16)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 30/125 (index 36)
INFO:kaldo:Completed IBZ k-point 30/125 (index 36)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 31/125 (index 2)
INFO:kaldo:Completed IBZ k-point 31/125 (index 2)
2026-07-19 08:20:25,080 - kaldo - INFO - Completed IBZ k-point 32/125 (index 48)
INFO:kaldo:Completed IBZ k-point 32/125 (index 48)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 33/125 (index 60)
INFO:kaldo:Completed IBZ k-point 33/125 (index 60)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 34/125 (index 30)
INFO:kaldo:Completed IBZ k-point 34/125 (index 30)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 35/125 (index 67)
INFO:kaldo:Completed IBZ k-point 35/125 (index 67)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 36/125 (index 115)
INFO:kaldo:Completed IBZ k-point 36/125 (index 115)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 37/125 (index 90)
INFO:kaldo:Completed IBZ k-point 37/125 (index 90)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 38/125 (index 105)
INFO:kaldo:Completed IBZ k-point 38/125 (index 105)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 39/125 (index 18)
INFO:kaldo:Completed IBZ k-point 39/125 (index 18)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 40/125 (index 19)
INFO:kaldo:Completed IBZ k-point 40/125 (index 19)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 41/125 (index 71)
INFO:kaldo:Completed IBZ k-point 41/125 (index 71)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 42/125 (index 39)
INFO:kaldo:Completed IBZ k-point 42/125 (index 39)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 43/125 (index 1)
INFO:kaldo:Completed IBZ k-point 43/125 (index 1)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 44/125 (index 58)
INFO:kaldo:Completed IBZ k-point 44/125 (index 58)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 45/125 (index 103)
INFO:kaldo:Completed IBZ k-point 45/125 (index 103)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 46/125 (index 57)
INFO:kaldo:Completed IBZ k-point 46/125 (index 57)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 47/125 (index 92)
INFO:kaldo:Completed IBZ k-point 47/125 (index 92)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 48/125 (index 109)
INFO:kaldo:Completed IBZ k-point 48/125 (index 109)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 49/125 (index 15)
INFO:kaldo:Completed IBZ k-point 49/125 (index 15)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 50/125 (index 38)
INFO:kaldo:Completed IBZ k-point 50/125 (index 38)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 51/125 (index 112)
INFO:kaldo:Completed IBZ k-point 51/125 (index 112)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 52/125 (index 94)
INFO:kaldo:Completed IBZ k-point 52/125 (index 94)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 53/125 (index 9)
INFO:kaldo:Completed IBZ k-point 53/125 (index 9)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 54/125 (index 96)
INFO:kaldo:Completed IBZ k-point 54/125 (index 96)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 55/125 (index 116)
INFO:kaldo:Completed IBZ k-point 55/125 (index 116)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 56/125 (index 123)
INFO:kaldo:Completed IBZ k-point 56/125 (index 123)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 57/125 (index 98)
INFO:kaldo:Completed IBZ k-point 57/125 (index 98)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 58/125 (index 124)
INFO:kaldo:Completed IBZ k-point 58/125 (index 124)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 59/125 (index 114)
INFO:kaldo:Completed IBZ k-point 59/125 (index 114)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 60/125 (index 83)
INFO:kaldo:Completed IBZ k-point 60/125 (index 83)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 61/125 (index 104)
INFO:kaldo:Completed IBZ k-point 61/125 (index 104)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 62/125 (index 84)
INFO:kaldo:Completed IBZ k-point 62/125 (index 84)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 63/125 (index 12)
INFO:kaldo:Completed IBZ k-point 63/125 (index 12)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 64/125 (index 110)
INFO:kaldo:Completed IBZ k-point 64/125 (index 110)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 65/125 (index 27)
INFO:kaldo:Completed IBZ k-point 65/125 (index 27)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 66/125 (index 100)
INFO:kaldo:Completed IBZ k-point 66/125 (index 100)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 67/125 (index 74)
INFO:kaldo:Completed IBZ k-point 67/125 (index 74)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 68/125 (index 95)
INFO:kaldo:Completed IBZ k-point 68/125 (index 95)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 69/125 (index 91)
INFO:kaldo:Completed IBZ k-point 69/125 (index 91)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 70/125 (index 106)
INFO:kaldo:Completed IBZ k-point 70/125 (index 106)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 71/125 (index 82)
INFO:kaldo:Completed IBZ k-point 71/125 (index 82)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 72/125 (index 68)
INFO:kaldo:Completed IBZ k-point 72/125 (index 68)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 73/125 (index 113)
INFO:kaldo:Completed IBZ k-point 73/125 (index 113)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 74/125 (index 21)
INFO:kaldo:Completed IBZ k-point 74/125 (index 21)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 75/125 (index 33)
INFO:kaldo:Completed IBZ k-point 75/125 (index 33)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 76/125 (index 111)
INFO:kaldo:Completed IBZ k-point 76/125 (index 111)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 77/125 (index 13)
INFO:kaldo:Completed IBZ k-point 77/125 (index 13)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 78/125 (index 14)
INFO:kaldo:Completed IBZ k-point 78/125 (index 14)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 79/125 (index 80)
INFO:kaldo:Completed IBZ k-point 79/125 (index 80)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 80/125 (index 24)
INFO:kaldo:Completed IBZ k-point 80/125 (index 24)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 81/125 (index 7)
INFO:kaldo:Completed IBZ k-point 81/125 (index 7)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 82/125 (index 64)
INFO:kaldo:Completed IBZ k-point 82/125 (index 64)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 83/125 (index 87)
INFO:kaldo:Completed IBZ k-point 83/125 (index 87)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 84/125 (index 49)
INFO:kaldo:Completed IBZ k-point 84/125 (index 49)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 85/125 (index 78)
INFO:kaldo:Completed IBZ k-point 85/125 (index 78)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 86/125 (index 117)
INFO:kaldo:Completed IBZ k-point 86/125 (index 117)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 87/125 (index 97)
INFO:kaldo:Completed IBZ k-point 87/125 (index 97)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 88/125 (index 6)
INFO:kaldo:Completed IBZ k-point 88/125 (index 6)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 89/125 (index 40)
INFO:kaldo:Completed IBZ k-point 89/125 (index 40)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 90/125 (index 61)
INFO:kaldo:Completed IBZ k-point 90/125 (index 61)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 91/125 (index 29)
INFO:kaldo:Completed IBZ k-point 91/125 (index 29)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 92/125 (index 63)
INFO:kaldo:Completed IBZ k-point 92/125 (index 63)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 93/125 (index 8)
INFO:kaldo:Completed IBZ k-point 93/125 (index 8)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 94/125 (index 47)
INFO:kaldo:Completed IBZ k-point 94/125 (index 47)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 95/125 (index 121)
INFO:kaldo:Completed IBZ k-point 95/125 (index 121)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 96/125 (index 79)
INFO:kaldo:Completed IBZ k-point 96/125 (index 79)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 97/125 (index 118)
INFO:kaldo:Completed IBZ k-point 97/125 (index 118)
2026-07-19 08:20:25,081 - kaldo - INFO - Completed IBZ k-point 98/125 (index 73)
INFO:kaldo:Completed IBZ k-point 98/125 (index 73)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 99/125 (index 70)
INFO:kaldo:Completed IBZ k-point 99/125 (index 70)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 100/125 (index 23)
INFO:kaldo:Completed IBZ k-point 100/125 (index 23)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 101/125 (index 42)
INFO:kaldo:Completed IBZ k-point 101/125 (index 42)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 102/125 (index 122)
INFO:kaldo:Completed IBZ k-point 102/125 (index 122)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 103/125 (index 108)
INFO:kaldo:Completed IBZ k-point 103/125 (index 108)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 104/125 (index 44)
INFO:kaldo:Completed IBZ k-point 104/125 (index 44)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 105/125 (index 26)
INFO:kaldo:Completed IBZ k-point 105/125 (index 26)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 106/125 (index 22)
INFO:kaldo:Completed IBZ k-point 106/125 (index 22)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 107/125 (index 76)
INFO:kaldo:Completed IBZ k-point 107/125 (index 76)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 108/125 (index 10)
INFO:kaldo:Completed IBZ k-point 108/125 (index 10)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 109/125 (index 56)
INFO:kaldo:Completed IBZ k-point 109/125 (index 56)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 110/125 (index 88)
INFO:kaldo:Completed IBZ k-point 110/125 (index 88)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 111/125 (index 17)
INFO:kaldo:Completed IBZ k-point 111/125 (index 17)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 112/125 (index 66)
INFO:kaldo:Completed IBZ k-point 112/125 (index 66)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 113/125 (index 54)
INFO:kaldo:Completed IBZ k-point 113/125 (index 54)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 114/125 (index 86)
INFO:kaldo:Completed IBZ k-point 114/125 (index 86)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 115/125 (index 62)
INFO:kaldo:Completed IBZ k-point 115/125 (index 62)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 116/125 (index 0)
INFO:kaldo:Completed IBZ k-point 116/125 (index 0)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 117/125 (index 20)
INFO:kaldo:Completed IBZ k-point 117/125 (index 20)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 118/125 (index 93)
INFO:kaldo:Completed IBZ k-point 118/125 (index 93)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 119/125 (index 11)
INFO:kaldo:Completed IBZ k-point 119/125 (index 11)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 120/125 (index 35)
INFO:kaldo:Completed IBZ k-point 120/125 (index 35)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 121/125 (index 50)
INFO:kaldo:Completed IBZ k-point 121/125 (index 50)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 122/125 (index 28)
INFO:kaldo:Completed IBZ k-point 122/125 (index 28)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 123/125 (index 65)
INFO:kaldo:Completed IBZ k-point 123/125 (index 65)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 124/125 (index 102)
INFO:kaldo:Completed IBZ k-point 124/125 (index 102)
2026-07-19 08:20:25,082 - kaldo - INFO - Completed IBZ k-point 125/125 (index 4)
INFO:kaldo:Completed IBZ k-point 125/125 (index 4)
2026-07-19 08:20:25,203 - kaldo - INFO - '_project_crystal' 7.12 s
INFO:kaldo:'_project_crystal' 7.12 s
The mean free path \(\lambda_\nu = v_\nu / \Gamma_\nu\) combines group velocity and scattering rate. Modes with long mean free paths are the dominant heat carriers.
mfp_norm = np.linalg.norm(conductivity.mean_free_path, axis=1)
plotter.plot_vs_frequency(phonons, mfp_norm, "mean free path")
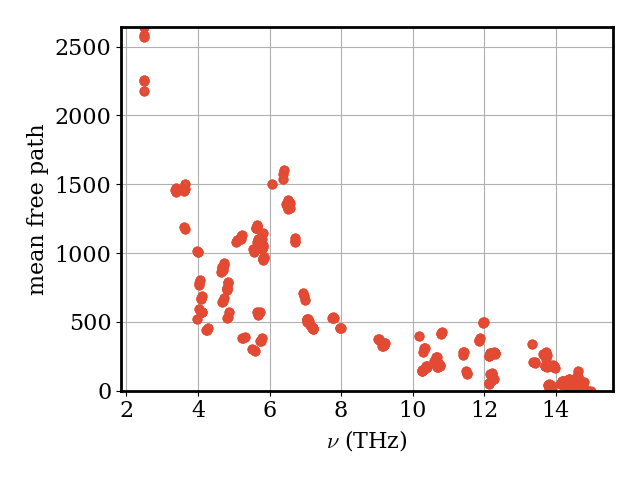
Finally, the thermal-conductivity tensor \(\kappa_{\alpha\beta}\) and its scalar average \(\bar\kappa = \tfrac{1}{3}\mathrm{Tr}\,\kappa\):
kappa = conductivity.conductivity.sum(axis=0) # sum over modes → (3, 3)
kappa_scalar = np.trace(kappa) / 3.0
print("Thermal conductivity tensor [W/(m·K)]:")
print(kappa)
print(f"\nScalar average: {kappa_scalar:.1f} W/(m·K)")
2026-07-19 08:20:26,655 - kaldo - INFO - Conductivity calculated
INFO:kaldo:Conductivity calculated
Thermal conductivity tensor [W/(m·K)]:
[[98.05140357 -0.98430438 -0.87002415]
[-1.18481573 98.98756595 -1.1848434 ]
[-0.8700886 -0.98382169 98.04944676]]
Scalar average: 98.4 W/(m·K)
We can also visualize how the cumulative thermal conductivity builds up as a function of phonon frequency, showing which frequency ranges contribute most to heat transport.
frequency = phonons.frequency.flatten() # THz
kappa_contrib = conductivity.conductivity.reshape(-1, 3, 3)
kappa_trace = np.array([np.trace(k) / 3.0 for k in kappa_contrib])
sort_idx = np.argsort(frequency)
freq_sorted = frequency[sort_idx]
kappa_cumulative = np.cumsum(kappa_trace[sort_idx])
fig, ax = plt.subplots()
ax.plot(freq_sorted, kappa_cumulative)
ax.set_xlabel("Frequency (THz)")
ax.set_ylabel(r"Cumulative $\kappa$ [W/(m·K)]")
ax.set_title("Cumulative thermal conductivity")
plt.tight_layout()
plt.show()
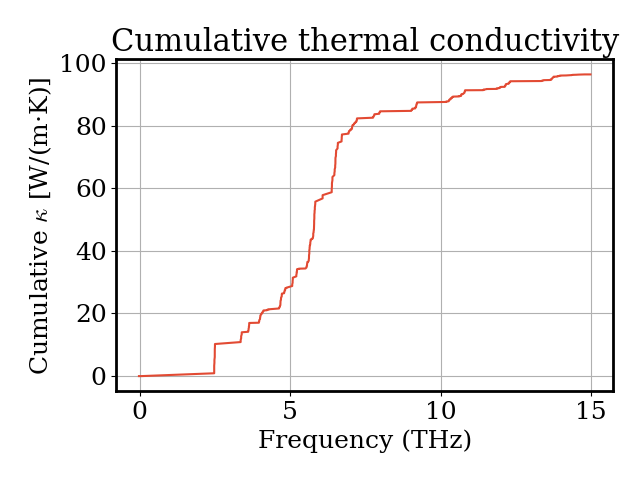
Convergence¶
The 3×3×3 supercells and 5×5×5 k-point mesh used above are chosen to keep CI runtime short and are not fully converged. To systematically converge \(\kappa\):
Increase the 2nd-order supercell until the phonon dispersion (especially the acoustic branches near \(\Gamma\)) is stable.
Increase the 3rd-order supercell until the scattering rates (and hence \(\kappa\)) converge.
Increase the k-point mesh until \(\kappa\) is converged with respect to Brillouin-zone sampling.
The pet-mad-xs model used here is faster but less accurate than
pet-mad-s; use the S model for production calculations.
More kALDo examples (different materials, advanced settings) are available in the kALDo examples repository.
Total running time of the script: (2 minutes 42.815 seconds)